

Hasar bölgelerinde biriken makrofajların, ksenobiyotiklere verilen patojenik yanıtta rol oynadığı görüşü 100 yıldan daha uzun bir süre önce Eli Metchnikoff tarafından ortaya konmuştur. İnflamatuar yanıtı “bazı zararlı etkilere karşı faydalı bir reaksiyon” olarak tanımlayarak, inflamasyon bölgesindeki hücreler tarafından salınan “fermentlerin” doku hasarına katkıda bulunabileceği hipotezini de öne sürmüştür (1). O zamandan bu yana bu kavramı destekleyen çok sayıda yayın yapılmıştır.
Bu yazımızda; akut ve kronik akciğer hasarında ve pulmoner toksik maddelerin neden olduğu hastalık patogenezinde makrofajların ve makrofajlar tarafından salınan inflamatuar medyatörlerin rolüne odaklanan bir derlemeden bahsedeceğiz.
İnflamatuar Makrofajlar
İnflamatuar makrofajlar, konak savunmasında ve zararlı uyaranlara karşı doğal bağışıklık sistemi yanıtında önemli bir rol oynayan mononükleer fagositlerdir. Embriyonik öncüllerden kaynaklanan yerleşik doku makrofajlarının (örn. alveolar makrofajlar) aksine, büyük ölçüde kan ve kemik iliği öncülerinden elde edilirler. CCR2 veya CX3CR1 gibi spesifik kemokin reseptörlerini eksprese ederler ve hasarlı hücre ve dokulardan salınan kemokinlere yanıt olarak dokularda birikirler (2-4). Doku hasarının olduğu bölgelerde lokalize olduktan sonra, bu makrofajlar, doku mikroçevresinde karşılaştıkları medyatörler tarafından aktive edilip değişen seviyelerde proinflamatuar/sitotoksik (M1) veya anti-inflamatuar/yara onarımı (M2) aktivitesi sergileyen alt sınıflara dönüşürler (5-7). Makrofajların M1 ve M2’ye dönüşümü sıkı bir şekilde kontrol edilen, spesifik sinyal yollarını, transkripsiyon faktörlerini ve posttranslasyonel düzenleyici bağlantıları içeren bir süreçtir (Tablo 1) (8, 9).
Tablo 1: Makrofaj Aktivasyonu ve Polarizasyonunun Regülatörleri
| Regülatör | M1 | M2 |
| Ekstraselüler | ||
| Sitokinler | IL-1β, IL-6, IL-12,
IL-23, IFNγ, TNFα, MIP-1α |
IL-4, IL-13, IFNα,
IL-1 RA |
| Büyüme Faktörleri | GM-CSF | M-CSF, TGFβ |
| Eikozanoidler/ biyoaktif lipidler | LTB4; 12-HETE,
5- HETE |
Lipoxins, Resolvins,
Thromboxane, PGI2 |
| TLR-4 agonistleri | DAMPs, PAMPs,
LPS |
|
| NOD agonistleri | Peptidoglycans | |
| İntraselüler | ||
| Oksidatif stres | ROS, RNS | |
| Metabolizma | Anaerobik glikoliz, glikoz alımı, yağ asidi sentezi | Oksidatif glikoz metabolziması, yağ asidi oksidasyonu ve alımı |
| Sinyal yolakları | AKT2, NOTCH ½ | AKT1, RTKs |
| Nükleer | ||
| Transkripsiyon faktörleri | NFκB, AP-1,
STAT1, IRF1, IRF5, IRF8, HIF-1α |
IRF4, KLF-4, c-myc,
PPARγ, RXRs, LXRs, STAT3, STAT6, IRF3, IRF4, HIF2α |
| Epigenetik | ||
| MikroRNA’lar ve histon modifikasyonları | miRNA-155,
miRNA-125b, miRNA-27b, miRNA-127, miRNA-223, miRNA-106a HDAC3 |
miRNA-146a/b
miRNA-21, miRNA-511-3p, miRNA-124, miRNA-125a/b, miRNA-24, miRNA-34a, let-7c |
| DNA metilasyonu | DNMT3b, DNMT1 | H3K27
demethylase DNMT3a, DNMT3al |
Proinflamatuar/sitotoksik M1 makrofajlar; tek başına interferon (IFN) γ‘ya yanıt olarak veya toll-like reseptör (TLR) 4 agonistleri (örn., lipopolisakkarit [LPS]; yüksek mobilite grup içeren protein ailesi üyesi olan HMGB1) ya da diğer sitokinler (örn., tümör nekroz faktörü [TNF] α ve granülosit makrofaj-koloni uyarıcı faktör [GM-CSF]) ile birlikte gelişirler.
Aktive M1 makrofajlar proinflamatuar sitokinleri (TNF α, interlökin (IL)-1β, IL-6, IL-12, IL-15, IL-23) salgılayıp, sitotoksik reaktif oksijen türlerini (ROS), reaktif nitrojen türlerini (RNS), proteolitik enzimleri ve biyoaktif lipidleri üretirler.
M1 makrofajlarının aktivitesi, inflamasyonu azaltan ve yara onarımını başlatan M2 makrofajları ile dengelenmiş durumdadır. Bu sürece, anti-inflamatuar sitokinler (IL-4, IL-10, IL-13), bazı eikosanoidler (lipoksinler, resolvinler) ve büyüme faktörleri (TGFβ, vasküler endotelyal büyüme faktörü [VEGF], epidermal büyüme faktörü [EGF], bağ dokusu büyüme faktörü [CTGF], fibroblast büyüme faktörü [FGF], trombosit kaynaklı büyüme faktörü [PDGF]) aracılık eder.
Akut akciğer hasarı ve persistan inflamasyon, M1 makrofajlarının uzun süreli veya aşırı bir immün yanıta ek olarak kusurlu M2 makrofaj aracılı doku onarımını içerirken, fibrozis ve kanser gibi kronik hastalıkların gelişiminin, M2 makrofajlarının alt gruplarının aşırı duyarlılıklarının bir sonucu olduğu düşünülmektedir.
M1 ve M2 makrofaj aktivasyonu oldukça dinamik bir süreçtir. Sinyal molekülleri, transkripsiyon faktörleri, epigenetik düzenleyiciler ve hücresel metabolizma patofizyolojik koşullara yanıt olarak değiştikçe, makrofajlar fenotiplerini ve işlevlerini kolayca değiştirirler. Örneğin başlangıçta proinflamatuar ve sitotoksik reaksiyonları kolaylaştıran hücreler, fenotipik değişime uğrayabilir ve böylece inflamasyon ve hasarın çözünmesinde rol oynayabilirler (10, 11). Sonuç olarak, makrofajların, proinflamatuar bir M1’den bir anti-inflamatuar/yara onarımı M2 fenotipine geçişi, normal yara iyileşmesi ve doku rejenerasyonunun ilerlemesi için çok kritik bir durumdur. Hipoksinin de M1’den M2’ye fenotipik geçişi bozduğuna dair bulgular söz konusudur (12).
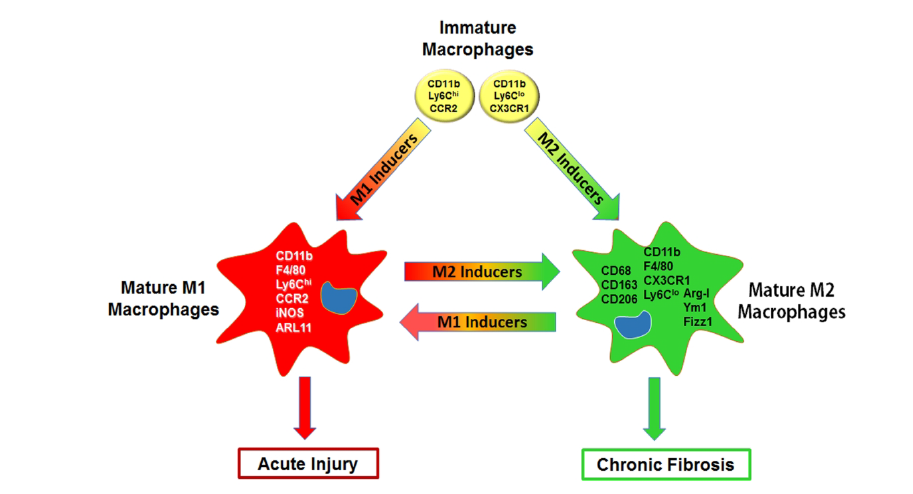
Şekil 1: Akut yaralanma ve fibroziste proinflamatuar/sitotoksik (M1) ve anti-inflamatuar/yara onarımı (M2) makrrofajlar. Olgunlaşmalarından pek çok faktör sorumludur ve indükleyen maddelere göre birbirlerine geçiş gösterebilirler.
Akut Akciğer Hasarı ve Onarımında İnflamatuar Makrofajlar
Akciğerde akut hasar, endotelyal ve epitelyal bariyerlerin bozulması ile ilişkilidir (13, 14). Proteinden zengin ödem sıvısının birikmesi, bronş epitelinin dökülmesi, nekrotik veya apoptotik Tip I hücrelerin görünümü, aşınmış bazal membran, genişlemiş ödematöz interstisyum, hasarlı endotelyal hücreler ve dokuda hücresel artıkların birikmesi ile karakterizedir. Hücresel özellikler, alveolar-kapiler membran bütünlüğünün kaybını, nötrofillerin ve makrofajların transepitelyal göçünü ve proinflamatuar/sitotoksik proteinlerdeki artışları içerir. Akut akciğer hasarının sonucu, toksik maddenin doğasına, maruz kalma dozuna, süresine ve spesifik doku konumuna bağlıdır. Bazı maruziyetlerden sonra akciğer yapısı ve fonksiyonu normale dönerken, diğer durumlarda, pulmoner vasküler yıkıma, fibrozan alveolite, çoklu organ yetmezliğine ve ölüme yol açan hasarın kalıcılığı veya ilerleyiciliği söz konusudur.
Akut akciğer hasarında makrofajların rolü olduğunu öne süren ilk kanıtlar, hayvanların çeşitli pulmoner toksik maddelere (asbest, radyasyon, bleomisin, endotoksin, silika, vb.) maruz kalmasının ardından dokuda bu hücrelerin sayısının arttığına dair bulgulara dayanmaktadır (6, 15-17). Ek olarak, pulmoner toksik maddeler tarafından indüklenen hasardan hemen sonra akciğerde biriken makrofajların, proinflamatuar/sitotoksik makrofajların prototipik göstergeleri olan; artan boyut ve vakuolizasyon, indüklenebilir nitrik oksit sentaz (iNOS) ve TNF α ekspresyonu dahil morfolojik değişikliklerle kanıtlandığı gibi, proinflamatuar bir M1 fenotipine doğru aktive olduğu görülmüştür. (6, 18-20).
Akut akciğer hasarında proinflamatuar/sitotoksik M1 makrofajlarının rolüne ilişkin doğrudan kanıt, doku hasarının makrofaj fonksiyonel durumu ile doğrudan ilişkili olduğu bulgularından gelir. Çeşitli yaklaşımların kullanıldığı bir dizi deneysel modelde (örneğin, farmakolojik, genetik veya makrofaj eksikliği olan fareler), makrofajları baskılayarak veya azaltarak pulmoner hasarın düzeldiği veya önlendiği gösterilmiştir. Örneğin, yapılan bazı çalışmalarda M1 makrofaj sitotoksik/proinflamatuar aktivite, anti-inflamatuar steroidler ile bloke edildiğinde, ozon, silika, bleomisin, hardal gazı, endotoksin, oleik asit veya hidrojen sülfürün neden olduğu akciğer hasarının azaldığı görülmüştür (6, 21-24). Benzer şekilde, M1 makrofaj aktivasyonunu engelleyen gadolinyum klorür ya da inflamatuar makrofajların sayısını azaltan klodronat lipozomları ön tedavi olarak uygulandığında akciğerde makrofaj birikimi ve ardından gelişen ozon, sigara dumanı, radyasyon ve silikanın toksik etkilerinin azaldığı görülmüştür (6, 25-28).Yapılan başka bir çalışmada, bleomisin kaynaklı akut akciğer hasarı ve iNOS ekspresyonu, M1 makrofajları oluşturamayan CCR4 eksik farelerde görülmemiştir (29). Radyasyon, ozon ve bleomisine yanıt olarak gelişen toksisitede artışlar; homeostatik koşullar altında akciğer proinflamatuar makrofaj aktivitesini baskılama işlevi gören pulmoner kolektin, sürfaktan protein D’den yoksun farelerde gözlenmiştir (20, 30, 31).
Akut hasarın çözülmesi, normal akciğer yapısına ve işlevine dönüş, aktif ve koordineli bir süreçtir. Kanıtlar, proinflamatuar medyatörlerin salınımını baskılayan ve doku onarım süreçlerini aktive eden karşı düzenleyici mekanizmaları uyaran M2 makrofajlarının büyük ölçüde bu süreçlere aracılık ettiğini göstermektedir. Hayvanların ozon, hardal gazı, bleomisin, radyasyon, endotoksin, sigara dumanı ve asbeste maruz kalmasının ardından akciğerde anti-inflamatuar/yara onarımı M2 makrofajlarının arttığı bildirilmiştir (6, 7, 20, 32, 33). Ancak ortaya çıkmaları, M1 makrofajlarına kıyasla daha geç olmaktadır.
Ozon, endotoksin, hardal gazı, dizel egzozu, asbest, silika veya hiperoksiye maruz kalmayı takiben akciğerdeki M2 makrofajlarındaki artışlar, IL-4, IL-10 ve IL-13’ün yukarı regülasyonu ile ilişkilidir (5, 6, 33). Bu sitokinler, proinflamatuar/sitotoksik medyatörlerin makrofaj üretimini azaltır ve hücre dışı matriks proteinlerinin ve yara iyileşmesinde önemli olan büyüme faktörlerinin oluşumunu uyarır (3, 4). Özellikle, M2 makrofaj gelişimi için anahtar olan IL-13’ün uygulanması, fareleri ölümcül endotoksemiden korurken, anti-IL-13 antikorları, bu farelerin hayatta kalmasını önemli ölçüde azaltır (34).
İnflamatuar Makrofajlar ve Pulmoner Fibrozis
Kronik pulmoner fibrozis, kollajen ve diğer hücre dışı matriks bileşenlerinin aşırı birikiminin bir sonucu olarak ortaya çıkan akciğer yapısının geri dönüşümsüz yıkımı ve remodelling ile karakterize bir dizi hastalığı ifade eder. Bu, solunum yollarında skar doku gelişimine ve nefes almada zorluğa yol açar. Kanıtlar, düzensiz yara onarımının pulmoner fibrozise katkıda bulunan kilit bir faktör olduğunu ve bunun, en azından kısmen, uzun süreli inflamasyon veya pnömonit sonrası M1 ve M2 makrofajlarının arasındaki dengesizliğe bağlı olduğunu göstermektedir (35-37). Bu dengesizlik, skar dokunun çözünmesini ve matriksin parçalanmasını destekleyen, M1 makrofajlar tarafından üretilen anti-fibrotik sitokinler (CXCL10) ve MMP’lerin azalması ve fibroproliferatif doku remodellingini indükleyen, M2 makrofajlar tarafından salınan profibrotik mediatörlerin (TGF β,CTG F) aşırı salınımı ile ilişkilidir. Fibrotik doku alanlarındaki M2 makrofajlarındaki artışlar, idiyopatik pulmoner fibrozis, KOAH ve kistik fibrozlu hastaların akciğerlerinde kaydedilmiştir; dahası, dokudaki bu hücrelerin sayısı, kötüleşen bir prognoz ile doğrudan ilişkilidir.
Hem insanlarda hem de kemirgenlerde fibrogenez sırasında akciğerde biriken M2 makrofajları, fibroblast proliferasyonunu ve kollajen sentezini uyaran bir dizi önemli profibrotik medyatörün (TGF β, CTG F ve CCL 18) ana kaynağı olarak tanımlanmıştır (38, 39). M2 makrofajlarının ayrıca TNF α, IL-1, IL-10, IL-13, IL-33, trombosit kaynaklı büyüme faktörü (PDG F), fibroblast büyüme faktörü (FGF) ve fibronektin salınımı yoluyla fibrozu arttırdığı düşünülmektedir ki bu mediatörlerin fibrotik akciğer hastalığı olan insanlarda ve hayvanlarda arttığı gösterilmiştir (3, 4). Pulmoner fibroz gelişiminde hiperaktif M2 makrofajlarının rolünü destekleyen en güçlü kanıt, fibrojenik toksik maddelere (örn. bleomisin, radyasyon, silika) yanıtın, IL-10 veya IL-13’ü aşırı eksprese eden hayvanlarda veya M2 makrofaj aktivasyonunu kolaylaştıran IL-33’ün egzojen uygulamasıyla şiddetlendiğine dair bulgulardan gelir (6, 8, 40). Aksine M-CSF’si olmayan veya M2 makrofajlarını tüketen klodronat lipozomları ya da anti-CSF1R antikoru ile tedavi edilen hayvanlarda kollajen üretimi ve fibroz azalır (41-43).
Akut Akciğer Hasarı ve Fibroziste Yerleşik Alveolar Makrofajlar
Makrofajların fenotipik ve işlevsel olarak farklı alt popülasyonları, sağlıklı bireylerin ve deney hayvanlarının akciğerleri boyunca lokalizedir; bu yerleşik doku makrofajları alveolar makrofajlar, interstisyel makrofajlar, plevral makrofajlar, intravasküler makrofajlar ve hava yolu makrofajları olarak tanımlanmıştır (6). En büyük ve en gelişmiş olanları, sürfaktanın geri dönüşümünde kilit rol oynayan alveolar makrofajlardır. Yerleşik alveolar makrofajlar ayrıca, patojenlere ve solunan toksik maddelere yanıt vermek için stratejik olarak yerleştirilmiş pulmoner bağışıklık savunmasının merkezindedir. Diğer yerleşik makrofajlar gibi, homeostazı sağlama, hasara ve enfeksiyona karşı koruma rollerinin anahtarı olan M2-benzeri fenotipe sahiptirler (36). Sağlıklı akciğerde, alveolar makrofajlar, alveolar epitelyum ve sürfaktan protein D, CD200, IL-10, TGF β ve bir transmembran glikoproteini MUC-1 gibi moleküller ile TLR4, CD200R gibi makrofaj reseptörleriyle etkileşimleri ile kontrollü bir ortamda bulunurlar (35). Alveolar makrofaj-epitel temasındaki bozukluklar, erken inflamatuar sinyalleşmede kritik olay olarak kabul edilir (36). Yerleşik alveolar makrofajların, zararlı uyaranlara karşı akut inflamatuar yanıtı tetiklemede rol oynadığı gösterilmiştir.
Zararlı uyaranlara maruziyeti takiben yerleşik alveolar makrofaj aktivasyonu, inflamazomlar (örn., NLRP3) yoluyla da meydana gelebilir. Bunlar, inflamasyonu tetikleyen IL-1 ve IL-18 salınımına sebep olan kaspaz-1’i oligomerize ve aktive eden sitozolik multiprotein kompleksleridir. Steril inflamasyonda NLRP3 aktivasyonu, kristallerin (örn., kolesterol, ürat), partiküllerin (örn., silika, titanyum) veya nanomateryallerin (örn., karbon nanotüpler) fagositozunu takiben meydana gelir (44, 45). Çalışmalar, NLRP3 inflamazomunun ozon, titanyum nanopartiküller, asbest, silika ve partikül madde tarafından indüklenen akut akciğer hasarına katkıda bulunduğunu göstermektedir (46, 47).
Yerleşik alveolar makrofajlardan salınan inflamazom ve inflamazom bağlantılı sitokinlerin astım, KOAH ve fibrozis gibi kronik akciğer hastalıklarının gelişiminde rol oynadığını gösteren veriler de mevcuttur (48). Bu göz önüne alındığında, akut pulmoner fibrotik değişiklikler, idiyopatik pulmoner fibrozlu hastalarda ve ayrıca asbest kaynaklı fibrozda alveolar makrofajlarda artan IL-1β ve IL-18 seviyeleri ile ilişkilidir (46). Sigara dumanı, asbest, silika, karbon nanotüpler, nanomateryaller ve bleomisin gibi fibrojenik toksik maddelerin tümünün, IL-1β salgılanmasına yol açan alveolar yerleşik makrofajlarında NLRP3 inflamazomunu doğrudan aktive ettiği gösterilmiştir (46). IL-1β, epitel hücrelerinin ve fibroblastların kollajen üreten miyofibroblastlara dönüşümünü, çoğalmasını ve aktivasyonunu tetikleyen TGF β salınımını uyarır (49). Son çalışmalar, bleomisin kaynaklı fibrozun başlangıcından hemen önce, yerleşik makrofajların tükenmesinin patolojinin şiddetini değiştirmediğini göstermiştir (50). Bu veriler, yerleşik makrofajların fibrogeneze katkısının, hastalık sürecinin erken evrelerinde daha belirgin olduğunu göstermektedir.
Yerleşik alveolar makrofajların toksik maruziyet veya enfeksiyonlardan sonra düzenlenmiş hücre ölümüne (piroptoz, nekroptoz, METosis) maruz kalmaya eğilimli olduklarını gösteren çalışmalar söz konusudur (51). Bu sıkı bir şekilde düzenlenmiş hücre ölümü yolları, yüksek oranda inflamatuar ve immünojeniktir. TLR4 ligandları (örneğin, LPS, HMGB1), RAGE aktivasyonu, IL-1β ve TNF α gibi inflamatuarlar ve sitokinler dahil olmak üzere çeşitli faktörler tarafından indüklenebilirler (52). Yerleşik alveolar makrofaj ölümünün, dolaşımdaki monositlerin ve nötrofillerin hasarlı alana ve enfeksiyon bölgelerine göçünü ve inflamatuar yanıtları başlatmada anahtar olduğu düşünülmektedir (53). Makrofaj ölümü ve inflamasyonun karşılıklı olarak birbirini etkilediği ve inflamasyonun artmasına neden olan bir oto-amplifikasyon döngüsü oluşturduğuna dair artan çalışmalar söz konusudur (54).
Sonuç
Solunum yolu dış ortamla doğrudan temas halinde olduğundan, solunan patojenlerin ve toksik maddelerin olumsuz etkilerine karşı özellikle hassastır. Makrofajlar, bu zararlı ksenobiyotiklere karşı temel bir konakçı bağışıklık savunma mekanizmasını temsil eder. Ancak, etkili konak savunması, yara iyileşmesi ve homeostazın tekrar sağlanması, makrofajların aktivitesinin dikkatli bir şekilde kontrol edilmesini gerektirir. Kontrol mekanizmaların yokluğunda, makrofajlar aşırı aktive olur ve bu da akut hasarın alevlenmesine veya kronik akciğer hastalığının gelişmesine neden olur. Bu, makrofajların tek bir homojen hücre popülasyonundan oluşmaması; daha ziyade benzersiz fenotipik ve fonksiyonel özelliklere sahip alt popülasyonlardan oluştuğu gerçeği ile daha da karmaşık bir hal alır. Ayrıca, fenotiplerini hızla değiştirme kapasitesine de sahiptirler. Karışık bir popülasyon içinde tek tek makrofajların tanımlanmasına ve karakterizasyonuna izin veren yaklaşımların (tek hücreli RNAseq veya western blotlama) kullanımı ve ayrıca patojenik süreç sırasında farklı zamanlarda makrofajların koşullu yıkımına izin veren yaklaşımların (Cre-lox) kullanımı, bu hücrelerin akciğer hastalığının gelişimindeki rolünü değerlendirmede özellikle değerli olacaktır.
Toksik maddelere yanıt olarak akciğer makrofajları ve epitel hücreleri arasındaki etkileşimlere odaklanmak da önemlidir. Bu bağlamda, hiperoksi veya toksik madde kaynaklı akciğer hasarını takiben oluşan epitelyal hücre kaynaklı mikroveziküllerin makrofajların proinflamatuar aktivasyonunda anahtar olduğunu gösteren son çalışmalar, yeni bir araştırma alanı oluşturmaktadır. Gelecekteki araştırmalar için başka bir ilgi alanı, pulmoner toksik maddelere karşı patojenik yanıtta ROS, proteazlar ve TNF α ‘ya yanıt olarak inflamatuar makrofajlardan salınan hücre dışı tuzakların (DNA ve proteinlerden oluşan lifler) rolüdür. Makrofaj aktivasyonunu düzenleyen yolakları ve salgıladıkları mediyatörleri anlamak, akciğer hastalıklarının ve solunan toksik maddelerin neden olduğu hastalıkların tedavisinde daha etkili yaklaşımlara yol açabilir.
KAYNAKLAR
- Steven JL. Metchnikoff on the Comparative Pathology of Inflammation. Glasgow Med J. 1892;38(3):195-205.
- Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117(4):902-9.
- Vannella KM, Wynn TA. Mechanisms of Organ Injury and Repair by Macrophages. Annu Rev Physiol. 2017;79:593-617.
- Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44(3):450-62.
- Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. 2014;14(2):81-93.
- Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macrophages and tissue injury: agents of defense or destruction? Annu Rev Pharmacol Toxicol. 2011;51:267-88.
- Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223(4-5):383-96.
- Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. 2014;5:614.
- Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11(11):750-61.
- Porcheray F, Viaud S, Rimaniol AC, Léone C, Samah B, Dereuddre-Bosquet N, et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142(3):481-9.
- Parisi L, Gini E, Baci D, Tremolati M, Fanuli M, Bassani B, et al. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J Immunol Res. 2018;2018:8917804.
- Faulknor RA, Olekson MA, Ekwueme EC, Krzyszczyk P, Freeman JW, Berthiaume F. Hypoxia Impairs Mesenchymal Stromal Cell-Induced Macrophage M1 to M2 Transition. Technology (Singap World Sci). 2017;5(2):81-6.
- Tam A, Wadsworth S, Dorscheid D, Man SF, Sin DD. The airway epithelium: more than just a structural barrier. Ther Adv Respir Dis. 2011;5(4):255-73.
- Müller-Redetzky HC, Suttorp N, Witzenrath M. Dynamics of pulmonary endothelial barrier function in acute inflammation: mechanisms and therapeutic perspectives. Cell Tissue Res. 2014;355(3):657-73.
- Bekki K, Ito T, Yoshida Y, He C, Arashidani K, He M, et al. PM2.5 collected in China causes inflammatory and oxidative stress responses in macrophages through the multiple pathways. Environ Toxicol Pharmacol. 2016;45:362-9.
- Hiraiwa K, van Eeden SF. Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediators Inflamm. 2013;2013:619523.
- Weinberger B, Laskin JD, Sunil VR, Sinko PJ, Heck DE, Laskin DL. Sulfur mustard-induced pulmonary injury: therapeutic approaches to mitigating toxicity. Pulm Pharmacol Ther. 2011;24(1):92-9.
- Sugiura H, Ichinose M. Nitrative stress in inflammatory lung diseases. Nitric Oxide. 2011;25(2):138-44.
- Aggarwal NR, King LS, D’Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol. 2014;306(8):L709-25.
- Malaviya R, Laskin JD, Laskin DL. Anti-TNFα therapy in inflammatory lung diseases. Pharmacol Ther. 2017;180:90-8.
- Huang B, Wang DX, Deng W. Protective effects of dexamethasone on early acute lung injury induced by oleic acid in rats. Int J Clin Exp Med. 2014;7(12):4698-709.
- Geng P, Ma T, Xing J, Jiang L, Sun H, Zhu B, et al. Dexamethasone ameliorates H(2)S-induced acute lung injury by increasing claudin-5 expression via the PI3K pathway. Hum Exp Toxicol. 2018;37(6):626-35.
- Wigenstam E, Rocksén D, Ekstrand-Hammarström B, Bucht A. Treatment with dexamethasone or liposome-encapsuled vitamin E provides beneficial effects after chemical-induced lung injury. Inhal Toxicol. 2009;21(11):958-64.
- Chen F, Gong L, Zhang L, Wang H, Qi X, Wu X, et al. Short courses of low dose dexamethasone delay bleomycin-induced lung fibrosis in rats. Eur J Pharmacol. 2006;536(3):287-95.
- Nemmar A, Nemery B, Hoet PH, Van Rooijen N, Hoylaerts MF. Silica particles enhance peripheral thrombosis: key role of lung macrophage-neutrophil cross-talk. Am J Respir Crit Care Med. 2005;171(8):872-9.
- Frank EA, Birch ME, Yadav JS. MyD88 mediates in vivo effector functions of alveolar macrophages in acute lung inflammatory responses to carbon nanotube exposure. Toxicol Appl Pharmacol. 2015;288(3):322-9.
- Poole JA, Gleason AM, Bauer C, West WW, Alexis N, van Rooijen N, et al. CD11c(+)/CD11b(+) cells are critical for organic dust-elicited murine lung inflammation. Am J Respir Cell Mol Biol. 2012;47(5):652-9.
- Pérez-Rial S, del Puerto-Nevado L, Terrón-Expósito R, Girón-Martínez Á, González-Mangado N, Peces-Barba G. Role of recently migrated monocytes in cigarette smoke-induced lung inflammation in different strain of mice. PLoS One. 2013;8(9):e72975-e.
- Trujillo G, O’Connor EC, Kunkel SL, Hogaboam CM. A novel mechanism for CCR4 in the regulation of macrophage activation in bleomycin-induced pulmonary fibrosis. Am J Pathol. 2008;172(5):1209-21.
- Casey J, Kaplan J, Atochina-Vasserman EN, Gow AJ, Kadire H, Tomer Y, et al. Alveolar surfactant protein D content modulates bleomycin-induced lung injury. American journal of respiratory and critical care medicine. 2005;172(7):869-77.
- Groves AM, Gow AJ, Massa CB, Laskin JD, Laskin DL. Prolonged injury and altered lung function after ozone inhalation in mice with chronic lung inflammation. Am J Respir Cell Mol Biol. 2012;47(6):776-83.
- Kambara K, Ohashi W, Tomita K, Takashina M, Fujisaka S, Hayashi R, et al. In vivo depletion of CD206+ M2 macrophages exaggerates lung injury in endotoxemic mice. Am J Pathol. 2015;185(1):162-71.
- Venosa A, Malaviya R, Choi H, Gow AJ, Laskin JD, Laskin DL. Characterization of Distinct Macrophage Subpopulations during Nitrogen Mustard-Induced Lung Injury and Fibrosis. Am J Respir Cell Mol Biol. 2016;54(3):436-46.
- Hong T, Li S, Guo X, Wei Y, Zhang J, Su X, et al. IL-13 Derived Type 2 Innate Lymphocytes Ameliorates Cardiomyocyte Apoptosis Through STAT3 Signaling Pathway. Front Cell Dev Biol. 2021;9:742662-.
- Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol. 2011;2:65.
- Alber A, Howie SEM, Wallace WAH, Hirani N. The role of macrophages in healing the wounded lung. Int J Exp Pathol. 2012;93(4):243-51.
- Byrne AJ, Maher TM, Lloyd CM. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol Med. 2016;22(4):303-16.
- Shvedova AA, Kisin ER, Mercer R, Murray AR, Johnson VJ, Potapovich AI, et al. Unusual inflammatory and fibrogenic pulmonary responses to single-walled carbon nanotubes in mice. Am J Physiol Lung Cell Mol Physiol. 2005;289(5):L698-708.
- Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, Goldmann T, et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med. 2006;173(7):781-92.
- Luzina IG, Kopach P, Lockatell V, Kang PH, Nagarsekar A, Burke AP, et al. Interleukin-33 potentiates bleomycin-induced lung injury. Am J Respir Cell Mol Biol. 2013;49(6):999-1008.
- Baran CP, Opalek JM, McMaken S, Newland CA, O’Brien JM, Jr., Hunter MG, et al. Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176(1):78-89.
- Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. 2011;184(5):569-81.
- Meziani L, Mondini M, Petit B, Boissonnas A, Thomas de Montpreville V, Mercier O, et al. CSF1R inhibition prevents radiation pulmonary fibrosis by depletion of interstitial macrophages. Eur Respir J. 2018;51(3).
- Nakayama M. Macrophage Recognition of Crystals and Nanoparticles. Front Immunol. 2018;9:103.
- Hosseinian N, Cho Y, Lockey RF, Kolliputi N. The role of the NLRP3 inflammasome in pulmonary diseases. Ther Adv Respir Dis. 2015;9(4):188-97.
- Sayan M, Mossman BT. The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases. Part Fibre Toxicol. 2016;13(1):51.
- Michaudel C, Couturier-Maillard A, Chenuet P, Maillet I, Mura C, Couillin I, et al. Inflammasome, IL-1 and inflammation in ozone-induced lung injury. Am J Clin Exp Immunol. 2016;5(1):33-40.
- Grailer JJ, Canning BA, Kalbitz M, Haggadone MD, Dhond RM, Andjelkovic AV, et al. Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol. 2014;192(12):5974-83.
- dos Santos G, Kutuzov MA, Ridge KM. The inflammasome in lung diseases. American journal of physiology Lung cellular and molecular physiology. 2012;303(8):L627-L33.
- Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol. 2013;49(4):503-10.
- Ginhoux F, Bleriot C, Lecuit M. Dying for a Cause: Regulated Necrosis of Tissue-Resident Macrophages upon Infection. Trends Immunol. 2017;38(10):693-5.
- Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21(8):1229-39.
- Fan EKY, Fan J. Regulation of alveolar macrophage death in acute lung inflammation. Respir Res. 2018;19(1):50.
- Linkermann A, Stockwell BR, Krautwald S, Anders HJ. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14(11):759-67.