Dr. Öğr. Üyesi Vildan Özer, Karadeniz Teknik Üniversitesi, Acil Tıp AD, KTÜ Tıbbi Toksikoloji Ünitesi Direktörü, Medical Toxicology Fellow at Emory University
Acil Tıp Uzmanı Dr. Özlem Bülbül, Giresun Devlet Hastanesi, Future Medical Toxicology Fellow at Emory University
YAZAR NOTU: Yazarlar makaleyi çevirirken çoğunlukla cümlenin tam karşılığını belirtmiş olsalar da cümlenin Türkçe çevirisinin anlamının bozuk olduğu durumlarda, daha anlaşılır bir Türkçe için çeviri değişikliği yapmışlardır. Ayrıca makalenin 2020 yılında yayınlanmış olması nedeniyle (The Crashing Toxicology Patient. Aaron Skolnik, Jessica Monas. Emerg Med Clin N Am. 38 (2020) 841–856, https://doi.org/10.1016/j.emc.2020.06.014), bazı toksikoloji bilgilerinde son 5 yıl içerisinde güncel değişiklikler olduğu görülmüştür.
Bu noktada yazarlar güncel bilgileri verebilmek için ilgili bölümlerde “YAZAR NOTU” başlığı altında en güncel ve en pratik bilgileri okuyucuya sunmayı hedeflemiştir. Yine makalenin yazarları ile karşı fikre düşülen durumlarda da bu başlık altında okuyucuya bilgi verilmesi hedeflenmiştir.
KRİTİK TOKSİKOLOJİ HASTASININ YÖNETİMİ
Anahtar Noktalar:
- Konvansiyonel antiepileptikler, ilaca bağlı nöbetleri veya status epileptikusu sonlandırmada tipik olarak inefektiftir.
- İlaçların sebep olduğu kardiyojenik şokun tedavisi, geleneksel şok tedavisinden farklıdır. Örneğin KKB ve Beta blokör gibi ilaçların zehirlenmelerine bağlı gelişen şok tedavisinde “hiperinsülinemik-öglisemik tedavi (HIET)” gibi antidotlar birinci basamak tedaviler arasında yer almaktadır.
- Zehirlenemeye bağlı gelişen ilaç kaynaklı ARDS, kardiyojenik şok veya kardiyak arrest durumunda, zehirlenmiş hastalara uygulanan ekstrakorporeal yaşam destek tedavileri (ECLS), ECLS’nin uygulandığı diğer endikasyonlara göre daha iyi bir sağ kalım oranına sahip olabilir. Bu nedenle zehirlenme ile başvuran ve ARDS, kardiyojenik şok veya kardiyak arrest gelişen hastalarda acil ECLS düşünülmelidir.
- İlaçlar ABD ve Avrupa’da akut KC yetmezliğinin önde gelen nedenidir. N-asetilsistein tedavisi, ilaç kaynaklı (Drug induced) karaciğer yetmezliğinden şüphelenilen tüm hastalara başlanmalı ve bu tür hastalar nakil yapabilen bir merkeze yönlendirilmelidir.
- Pek çok kritik derecede zehirlenmiş hasta, geleneksel olarak renal replasman terapisi için bir endikasyona sahip olabilir. İlaç veya toksinin uzaklaştırılması isteniyorsa, intermittant (aralıklı) hemodiyaliz, CRRT gibi sürekli terapilere göre daha üstün bir ilaç klirensi sağlar.
GİRİŞ
Acil tıp hekimleri, ilaç ve toksin ilişkili acil servis başvurularının rutin yönetimi konusunda yetkindir. Son zamanlarda, zehirlenme ilişkili acil servis başvurularında artış gözlenmektedir. Bu artışa paralel olarak; hastaların hastanede kalış süreleri uzamakta, klinik tabloları daha karmaşık hale gelmekte, kaynak kullanımı artmakta ve hastaneye yatış olasılıkları artmaktadır. 2017 yılında, Amerika Birleşik Devletleri Hastalık Kontrol ve Önleme Merkezi (CDC) tarafından ABD’de 75.354 zehirlenme ilişkili ölüm olgusu bildirilmiştir. Zehirlenme ile başvuran hastalar; genellikle hastalığın ilk birkaç saatinde en kritik dönemindedirler. Bu nedenle, acil tıp ve yoğun bakım uzmanları, kritik toksikoloji hastalarının tanısının konması ve bu hastaların tedavi yönetiminde birincil sorumluluğu taşımaktadırlar. Bu hastaların yönetiminde hastaların acil resüsitasyonu ve stabilizasyonu öncelikli olup, tanısal testler ve spesifik antidot uygulamaları ikincil öneme sahiptir.
NÖROLOJİK TOKSİSİTE
Nöbet ve Status Epileptikus:
İlaç kaynaklı nöbetler yaygın olarak görülmektedir. Bazı vaka serilerinde, status epileptikus vakalarının %9’unun ve yeni başlangıçlı nöbetlerin %6’sının nedeninin ilaç kaynaklı nöbetler olduğu bildirilmiştir. İlaç kaynaklı nöbetlerde status epileptikus gelişme riski daha yüksek olup vakaların %10 kadarı karmaşık hale gelebilir. İlaç kaynaklı nöbetler, ilaç ilişkisiz nöbetlerle karşılaştırıldığında; ilaç kaynaklı nöbetlerin, hipoksi, hiperkapni, rabdomiyoliz, metabolik asidoz, laktat artışı, aşırı metabolik ihtiyaca bağlı beyin hasarı gibi komplikasyonlarla daha sık ilişkili olduğu ve mortalite riskinin daha yüksek olduğu gösterilmiştir. Çoğu epilepsinin aksine, ilaç kaynaklı nöbetler genellikle yaygın bir beyin süreci olarak başlar ve sıklıkla inhibitör (gama-aminobütirik asit [GABA]) ve eksitatör (asetilkolin, glutamat, dopamin, norepinefrin ve serotonin) nörotransmitterlerin dengesinin bozulmasından kaynaklanır. Bu durum, GABAA reseptör antagonizması veya modülasyonu, GABAA veya GABABagonistlerinin kronik kullanımının kesilmesi, çekilmesi ya da aşırı eksitatör iletim ile ilişkili olabilir (Tablo 1). Bu nedenle, geleneksel antiepileptik ilaçlardan olan fenitoin, bu nöbetleri sonlandırmada genellikle etkisizdir.
İlaç kaynaklı nöbetlerin tedavisinde, öncelikle hasta hızla stabilize edilmeli ve SSS’deki inhibisyon-eksitasyon dengesinde eksitasyon yönüne kayan dengenin, inhibisyon ayağını artırarak denge yeniden sağlanmalıdır. Hastanın hava yolu değerlendirilmeli ve havayolu güvenliği sağlanmalı, parmak ucu kan şekeri, sodyum düzeyleri ölçülmeli veya bu testler yapılamıyorsa ampirik olarak dekstroz uygulanmalıdır. Hipotansiyon gelişen hastalarda, dengeli kristaloid solüsyonların ampirik uygulanması veya sıvıya yanıt vermeyen olgularda vazopressör kullanımı önerilir. Vücut core sıcaklığı ölçülmeli ve hipertermi saptanması durumunda aktif soğutma yöntemleri uygulanmalıdır. İlaç kaynaklı nöbetlerin tedavisinde birinci basamak ajanlarGABAA agonistleri ve benzodiazepinlerdir (Tablo 2). İzoniazid veya hidrazin (örneğin, Gyromitra esculenta [yanlış kuzugöbeği] mantarı zehirlenmesi) zehirlenmesinden şüpheleniliyorsa, piridoksin uygulanmalıdır. Eğer hasta status epileptikusdurumunda kalırsa, epilepsi tedavisinde kullanılan ikinci basamak ajanlara geçilmelidir. İkinci basamak ajanlar arasında fenobarbital, yüksek doz midazolam infüzyonu ve propofol yer almaktadır. Status epileptikus yönetiminde, burst supresyon sağlamak için gereken propofol dozu, yoğun bakım ünitesinde (YBÜ) sedasyon amacıyla kullanılan dozlardan daha yüksektir. Ketaminin umut vadeden etkileri olmasına rağmen, randomize kontrollü çalışmaların eksikliği nedeniyle tedavi algoritmasında rutin olarak önerilmemektedir.
Tablo 1: Zehirlenme ilişkili Nöbetler.
| MEKANİZMA | Neden olan ajanlar |
| GABAA reseptör antagonizması veya modülasyonu | FlumazenilCiprofloksasinKlozapinCicutoksin |
| GABAA veya GABAB agonistlerinin kronik kullanımının kesilmesi, çekilmesi | EtanolBenzodiazepinlerBarbitüratlarBaklofenGama Hidroksibütirat (GHB)Gama Bütirolakton (GBL) |
| Aşırı eksitatör iletim | SempatomimetiklerSeratonin sendromuMonoamin oksidaz inhibitörleri |
| GABA üretimi inhibisyonu | İzoniazidHidrazinGyromitra türü mantarlar |
| Adenozin antagonizması | KarbamazepinKafeinTeofilin |
| Tablo 2: İlaç ilişkili nöbetlerin tedavisi. | |
| Başlangıç Stabilizasyonu | Destekleyici bakım sağla Havayolu yönetimiHipotansiyon yönetimiDengeli Kristaloid mayi verSıvı yanıtı yoksa Vazopressör başlaHipertermiyi kontrol et ve yönet Parmak ucu kan şekeri ve bazal metabolik değerleri görüntüleAmpirik 25 gram IV dekstroz ver (Eğer bu tetkikleri yapamıyorsan) |
| Birinci Basamak Tedavi | Birinci basamak ajanları uygulaBenzodiazepinlerLorazepam, 4 mg, IV, her 4-5 dk’da bir veyaMidazolam, 5 mg, IMİzoniazid veya hidrazin zehirlenmesi şüphesi varsaPiridoksinYutulan izoniazid gramajına göre verilecek doz hesaplanabilir ya da 25 mg/kg, IV, 15-30 dk içinde (Max: 5 gr) |
| İkinci Basamak Tedavi | Henüz yapılmadıysa havayolu güvenliğini sağlaİkinci basamak ajanları uygulaFenobarbital IV veyaYüksek doz Midazolam veyaPropofol, 80 mcg/kg/dk |
| Tanısal Değerlendirme | Serum APAP ve Salisilat düzeyi, idrar toksikoloji paneli gönderEğer endikeyse antiepileptik ilaç düzeyi gönderBeyin BT planlaStatus epileptikus, uzamış postiktal durum veya kafa travması mevcutsaSürekli EEG monitörizasyonunu düşünStatus epileptikus mevcutsa |
| Yatış/Taburculuk Planlaması | Yoğun bakım yatışı |
Tanısal Değerlendirme: Nörotoksisite
Epilepsi tanısı olan hastalar, ilaçlarını reçete edildiği şekilde alırken bile ilaç kaynaklı nöbet geçirebilirler. Ayrıca, karbamazepin gibi bazı antiepileptik ilaçlar, supraterapötik dozlarda nöbete sebep olabilirler. Antiepileptik ilaç maruziyeti olan hastalarda ilaç düzeyleri mutlaka değerlendirilmelidir. İdrar toksikolojisi veya serumda ilaç taraması genellikle sınırlı bir tanısal değere sahiptir; ancak, sonuçların pozitif olması diğer tetkiklerin negatif olması durumunda, idrar toksikolojisi veya serumda ilaç taramasında elde edilen pozitif sonuçlar tanıya yardımcı olabilir. Asetaminofen ve salisilat gibi sık kullanılan toksinlerin kan düzeyleri ölçülmelidir, zira salisilatlar fatal nörotoksisiteye yol açabilir.Status epileptikus, uzamış postiktal dönem, kafa travması açısından öykü veya fizik muayene bulguları bulunan hastalarda kontrastsız beyin bilgisayarlı tomografi (BT) tetkiki gereklidir. Endotrakeal entübasyon sırasında nöromüsküler blokör kullanılmış olsa dahi, status epileptikus tablosu ile başvuran tüm hastalarda sürekli elektroensefalografi (EEG) monitörizasyonu yapılmalıdır, çünkü tedavi edilen jeneralize konvülsif status epileptikus olgularının %14’ü nonkonvülsif status epileptikusa dönüşebilir. İlaç kaynaklı status epileptikus veya karmaşık nöbet geçiren tüm hastalar yoğun bakım ünitesine yatırılmalıdır.
SOLUNUM YETMEZLİĞİ
Çok sayıda ilaç, çeşitli mekanizmalarla akut solunum yetmezliğine neden olabilir. Birçok SSS depresanı, santral hipoventilasyona yol açarak solunum yetmezliğine katkıda bulunur. Günümüzde, opioidler acil tıp hekimleri tarafından en sık karşılaşılan ilaç kaynaklı solunum yetmezliği nedenlerinden biri olarak sayılmaktadır. Hipoventilasyonu olan bir hastada opioid intoksikasyonu ayırıcı tanıda yer alıyorsa, naloksan uygulanmalıdır.Naloksan genel olarak güvenli bir ajan olmakla birlikte, nadiren ciddi pulmoner ve kardiyak komplikasyonlara yol açabilir. Bu komplikasyonlar, akut opioid antagonizmasına bağlı katekolamin seviyelerindeki hızlı artış ile ilişkili olabilir ve başlangıç (>0,4 mg) ve toplam (>4,4 mg) doz arttıkça insidansının arttığı görülmektedir. Bununla birlikte, fentanil analogları veya diğer sentetik opioidlerin neden olduğu doz aşımını tersine çevirebilmek için birden fazla nalokson uygulaması ve yüksek başlangıç dozları gerekebilir. Sedatif ajanların büyük bir kısmı da hiperkapnik solunum yetmezliğini indükleyebilir veya hava yolu koruyucu reflekslerin kaybına neden olabilir.
Organofosfatlı pestisitler veya sinir ajanları gibi nöromüsküler güçsüzlüğe sebep olan toksinler, hipoventilasyona, solunum sistemindeki sekresyonların temizlenmesinde yetersizliğe ve solunum arrestine neden olabilir. Ayrıca, bazı ilaçların (örneğin, salisilatlar, kalsiyum kanal blokerleri) yüksek dozda alımı akut respiratuar distres sendromuna (ARDS) yol açabilir. Kardiyojenik (örneğin, beta blokerler, kokain) ve nörojenik (örneğin, nalokson) pulmoner ödem de ilaçların sebep olduğu iyi tanımlanmış klinik tablolardandır.
Solunum yetmezliğinin tüm nedenleri öncelikle endotrakeal entübasyon ve akciğer koruyucu mekanik ventilasyon stratejileriyle destekleyici olarak yönetilmelidir. Toksikoloji hastalarında uzamış immobilizasyona bağlı komplikasyonlar yaygın olup akut böbrek hasarı sık görüldüğünden, hızlı seri entübasyon (RSI) için nondepolarizan nöromüsküler blokerlerin tercih edilmesi önerilmektedir. Refrakter hipoksemik veya hiperkapnik solunum yetmezliği vakalarında, venovenöz ekstrakorporeal membran oksijenasyonu (V-V ECMO) ile yüksek sağkalım oranları bildirilmiştir.
KARDİYOVASKÜLER TOKSİSİTE
İlaç Kaynaklı Taşikardi ve Malign Aritmiler:
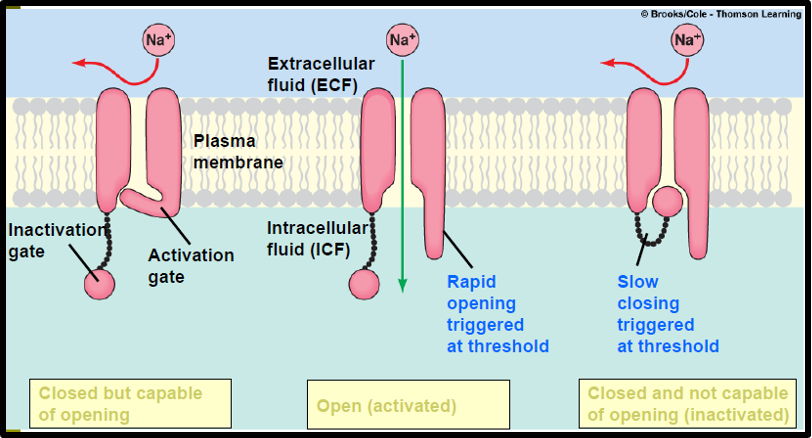
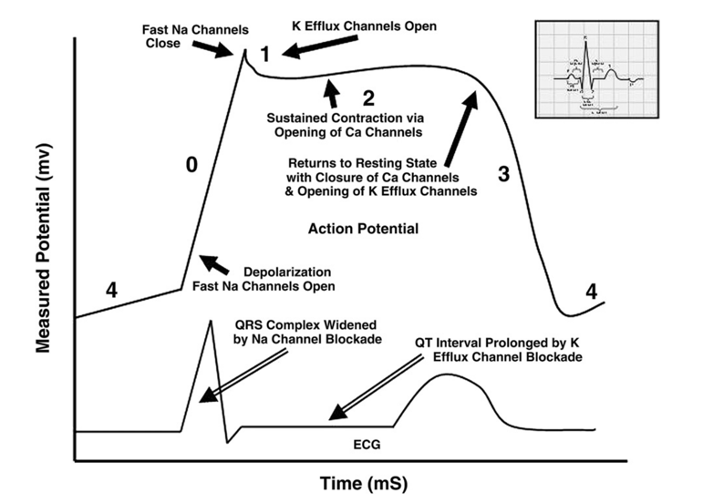
Sempatomimetik veya antikolinerjik zehirlenmeler sinüs taşikardisine neden olabilir. Klasik olarak bu iki tablo, antikolinerjik hastalarda mukozalarda ve aksillada (koltuk altında) kuruluk varlığı ile ayırt edilir. Periferik vazodilatasyona yanıt olarak gelişen refleks taşikardi nadir görülmekle birlikte, dihidropiridin grubu kalsiyum kanal blokerleri ile oluşan zehirlenmeler bu durumun istisnasıdır. Birçok ilaç, malign taşidisritmi riskini artırabilir. Sodyum kanal blokajına bağlı QRS genişlemesi, potasyum akışının blokajına bağlı QTc uzaması ve torsades de pointes gelişimi, sempatomimetik aktivite sonucu miyokard uyarılabilirliğinde artış ve miyokardın endojen katekolaminlere duyarlılığının artması gibi nedenler taşidisritminin yaygın mekanizmaları arasında yer almaktadır. Zehir danışma merkezinin retrospektif verilerine dayanan bir incelemede, taşidisritmi ile ilişkili ajanlar Tablo 3’te sunulmuştur. Bilinmeyen bir nedene bağlı nabızsız disritmi gelişen hastalarda, ilk tedavi yaklaşımı Spontan Dolaşımı Geri Döndürme (Return of Spontaneous Circulation, ROSC) amacıylaİleri Kardiyak Yaşam Desteği (Advanced Cardiac Life Support, ACLS) protokollerine uygun şekilde yürütülmelidir.
Tablo 3: Zehirlenme ilişkili disritmiler.
| KARDİYAK DÜZENSİZLİK | Neden olan ajanlar |
| Sinüs Taşikardisi | SempatomimetiklerAntikolinerjikler |
| Geniş QRS kompleksli Taşikardi | TCAStimülanlar (Kokain)DifenhidraminSitalopramPropoksifenBupropiyonLityumLamotrijinAntiaritmikler |
| Torsades de pointes | Siklik antidepresanlarMetadonAntipsikotiklerAntiaritmikler |
| Bradikardi / AV Blok | KKBBeta BlokörlerKardiyak glikozitler (örneğin digoksin)Organofosfat veya Karbamatlar (örneğin Sinir ajanları, Malathion, Pridostigmin)Santral α-2 agonistleri (Klonidin, Guanfasin) |
İlaç Kaynaklı Bradikardi:
Farklı derecelerde atriyoventriküler iletim bloklarını içeren ilaç kaynaklı bradikardi, çeşitli ilaç gruplarına bağlı olarak gelişebilir. En sık ölümle sonuçlanan kardiyak ilaç zehirlenmelerinin başlıca sorumluları kalsiyum kanal antagonistleri ve beta-adrenerjik antagonistlerdir. Diyabetik olmayan hastalarda hipotansiyon ve bradikardi ile birlikte belirgin hiperglisemi varlığı, kalsiyum kanal blokeri zehirlenmesini düşündürmelidir.Bradikardiye neden olduğu bilinen ajanlar Tablo 3’te sunulmuştur. İlaç kaynaklı oluşan semptomatik bradikardi, İleri Kardiyak Yaşam Desteği protokollerine göre yönetilir ve atropine yanıt verebilir. İntravenöz (IV) bolus veya infüzyon yoluyla uygulanan kalsiyum, inotropiyi ve kan basıncını artırabilir, ancak hayvan modellerinde bradikardi üzerine etkili olmadığı gösterilmiştir; insan olgularında ise sonuçlar değişkenlik göstermektedir. Toksikolojik vakalardaki bradikardi tedavisinde transkütan ve transvenöz kardiyak pacemaker uygulamaları denenmiş olsa da elektriksel capture’ın her zaman güvenilir olmadığı bildirilmiştir.Önemli disritmi, hiperkalemi (>6 mmol/L) veya hemodinamik instabilitesi olan vakalarda kardiyak glikozit zehirlenmesi düşünülüyor veya biliniyorsa, digoksin Fab fragmanları Tablo 4’te belirtilen şekilde uygulanabilir.
Sodyum Bikarbonat
İlaçlara bağlı sodyum kanal blokajı ve buna bağlı gelişen geniş kompleks taşikardi, birçok farklı ajanla ilişkilendirilmiştir(bkz. Tablo 3). Bu durumda kullanılacak birinci basamak antidot sodyum bikarbonattır, ancak sodyum bikarbonatın bu blokajı nasıl tersine çevirdiği tam olarak anlaşılamamıştır. Geleneksel olarak, sodyum bikarbonat tedavisine başlama eşiği QRS süresine göre belirlenmiş olsa da tedaviye başlama noktasında görüş birliği tam olarak sağlanamamıştır. Normal QRS süresi genellikle 80 ila 100 milisaniye (ms) arasında değiştiğinden, ilaç kaynaklı olduğundan şüphelenilen geniş kompleks taşikardi vakalarında, QRS süresi 120 milisaniyeden uzun ise sodyum bikarbonat uygulanması ve ardından hızla yeni EKG çekilerek QRS süresinde daralma meydana gelip gelmediğinin izlenmesi önerilir. Eğer QRS süresinde daralma gözlenirse, kan pH’ını 7.45–7.55 aralığında tutacak şekilde sodyum bikarbonat infüzyonu devam ettirilmelidir.Hastalardan, pH kontrolü için seri arter kan gazı ölçümleri alınmalıdır. Sodyum bikarbonat tedavisinin en bilinen ve beklenen komplikasyonu hipokalemidir. Hipokalemi, alkalozu telafi etmek için potasyum iyonlarının hücre içine, hidrojen iyonlarınınsa hücre dışına kayması nedeniyle oluşur. Bu komplikasyon potasyum replasmanı ve seri elektrolit ölçüm takipleri yapılarak tedavi edilebilir.
YAZAR NOTU: Sodyum bikarbonat tedavisine başlama eşiği konusunda her ne kadar ortak bir karar birliğine henüz varılmamış olsa da QRS süresinin eşiği belirlenirken literatürdeki çalışmalardan elde edilen bilgiler göz önünde bulundurulmaktadır. Bu raporlardan en çarpıcı olanı Boenhart ve Lovejoy tarafından yapılan çalışmadır. Bu çalışmada, QRS genişliği 100 ms’den büyük olan hastalarda nöbet insidansının %33 olduğu bulunmuştur. Ek olarak, QRS süresi 160 ms’den büyük olan hastalarda disritmi insidansı %50 bulunmuştur. Ancak, hiçbir hastada sırasıyla bu eşiklerin altında nöbet veya disritmi görülmemiştir. Bu sebeple güncel tıp pratiğinde QRS süresi 120 ms’yi aştığı zaman değil, 100ms’yi aştığı anda sodyum bikarbonat tedavisine başlanması, hastaların nöbet dahil hiçbir komplikasyonla karşılaşmaması için daha mantıklı görülmektedir. Elde edilen verilerden yola çıkarak hastaların QRS genişliğinin sebep olabileceği en az komplikasyonla karşılaşmaları için en güvenli sınırda kalmak tercih edilmektedir. Tüm bu sebeplerden dolayı normal bir hastada QRS süresinin 120 ms’yi aşması patolojik olarak kabul edilirken, toksikoloji hastalarında QRS süresinin 100 ms’yi aşması patolojik kabul edilir ve sodyum bikarbonat tedavisine başlanması için eşik değer olarak kabul edilmektedir. Bu yüzden yazar, çevrilen bu makalede önerilenden farklı bir QRS süresi eşik değerini önermektedir. Bunun dışında, başlanan sodyum bikarbonat infüzyonuna ne kadar süre devam edilmesi gerektiği konusunda da bir fikir birliğine varılmamıştır. Literatürde infüzyonun 24 saat devam etmesi gerektiğini belirten yazılar bulunmakla beraber genel uygulama hastanın en az 6 saat infüzyon alması ve infüzyon bitiminden sonra en az 24 saat monitörize şekilde takip edilmesidir (referans A).
Lipid Emülsiyon
İntravenöz lipid emülsiyonu (ILE) ilk olarak lokal anestezik toksisitesinin tedavisinde kullanılmıştır. Konsantre lipid solüsyonlarının (genellikle %20’lik çözelti) büyük miktarlarda uygulanması, hayvan modellerinde ve insan olgu bildirimlerinde lokal anesteziklerin kardiyovasküler toksisitesini tersine çevirmiştir. Daha sonra, ILE’nin trisiklik antidepresanlar, kalsiyum kanal blokerleri ve beta blokerler de dahil olmak üzere birçok farklı ilaç doz aşımında kurtarma tedavisi olarak kullanıldığı bildirilmiştir.Lipid emülsiyonunun hemodinami üzerindeki olumlu etkileri için çeşitli mekanizmalar öne sürülmüştür:
- Lipit havuzu (“lipid sink”) hipotezi, lipofilik ilaçların bu faza tercihli olarak dağılmasını sağlar. (Lipofilik ilaçların intravenöz olarak verilen lipid havuzuna hapsedilerek dolaşımdan uzaklaştırılmasını öne süren bir mekanizmadır)
- Kasılma fonksiyonu bozulmuş miyokard dokusu için serbest yağ asidi kaynağı sağlayarak kalp metabolizmasını destekler.
- Endotelyal nitrik oksit sentazın inhibisyonu yoluyla vazodilatasyonu azaltabilir.
İlaç aşırı dozu olan hastalarda ILE kullanımı yanında hayatı tehdit etmeyen doz aşımı vakalarında da ILE kapsamının genişletilebileceği söyleniyor olsa da yakın zamanda yapılan sistematik bir derlemede hayatı tehdit etmeyen doz aşımı vakalarındaki ILE etkinliğinin heterojen olduğu bildirilmiştir.Bununla birlikte, ILE tedavisi ile ilişkili birçok komplikasyon bildirilmiştir: Bunlar aşağıda listelenmiştir:
- Lipemiye bağlı hatalı laboratuvar test sonuçları
- Akut böbrek hasarı
- Kardiyak arrest
- Ventilasyon/perfüzyon uyumsuzluğu
- ARDS
- Venöz tromboemboli
- Hipersensitivite reaksiyonları
- Yağ embolisi veya lipid aşırı yüklenme sendromu
- Pankreatit
- Alerjik reaksiyonlar
- Artmış enfeksiyon duyarlılığı
Venoarteriyel ekstrakorporeal membran oksijenasyonu (VA-ECMO) uygulanan hastalarda, lipid emülsiyonunun ECMO devresinde aglütinasyon (tortu) oluşumuna, ECMO devresinde kullanılan akış musluklarının kırılıp çatlanmasına, ECMO devresinde bulunan oksijenatör yağ emülsiyon filtrelerinin tıkanmasına ve ECMO devrelerinde pıhtı oluşumuna sebep olduğu ve/veya bunların insidansında artışa neden olduğu bildirilmiştir. The American Academy of Clinical Toxicology’nin kanıta dayalı önerileri, ILE’nin bupivakain toksisitesine bağlı kardiyak arrest tedavisinde kullanılmasını desteklemekte, ancak diğer çoğu zehirlenme için ilk basamak tedavi olarak kullanılmasını önermemektedir. Alternatif tedaviler başarısız olduğunda, bupivakain toksisitesinde ILE kullanımı önerilmekte; diğer lokal anestezikler, amitriptilin ve bupropion toksisitelerinde ise kullanımı sadece tavsiye edilmektedir. Diğer toksinler için ise öneriler tarafsız kalmaktadır. İleri tedavi seçeneklerinin (örneğin mekanik dolaşım desteği) hızla erişilebilir olmadığı durumlarda ve diğer antidotal tedavilerin başarısız olduğu ilaç kaynaklı kardiyak arrest vakalarında lipid emülsiyonu bir tedavi seçeneği olarak düşünülebilir.
| Tablo 4: Sık kullanılan antidotların dozları. | |||
| ANTİDOT | ENDİKASYON | BOLUS | İNFÜZYON |
| İntravenöz NAC | Asetaminofen zehirlenmesi (nomograma göre 4. Saat APAP düzeyi eşik değerin üzerinde ise)Akut KC yetmezliği | 150 mg/kg 1 saatte yükleme dozu, ardından: | 12,5 mg/kg/h 4 saat boyunca, ardından 6,25 mg/kg/h 16 saat boyunca IV infüzyon. |
| YAZAR NOTU: Parasetamol zehirlenmelerinde kullanılan IV NAC protokolü farklı dozlardaki NAC infüzyonunun 3 farklı torbada verilmesi ve toplamda 21 saatlik bir infüzyon esasına dayanır. Ancak yeni çalışmalar daha az yan etki profiline sahip olması ve 3 torba rejimiyle aynı etkinlikte hepatotoksisiteden koruması nedeniyle 2 torba rejimini önermektedir. Yeni güncellenen rejime göre 2 torba NAC protokolü: yükleme dozu olmadan 200 mg/kg 4 saatte infüzyon (birinci torba), sonrasında 100 mg/kg 16 saatte infüzyon (ikinci torba), toplamda 20 saatlik IV infüzyon şeklindedir (referans B). | |||
| Glukagon | Miyokardiyal disfonksiyon gelişen Beta Blokör toksisitesi | 50-150 mcg/kg, 10 mg’a kadar IV | Yanıt alınan dozda infüzyona başlanmalıdır.Örneğinetkili bolus dozu 5 mg ise, ardından 5 mg/h hızında infüzyon başlanmalıdır. |
| YAZAR NOTU: Beta bloker toksisitesinde intravenöz (IV) glukagon uygulaması önerilen bir tedavi yaklaşımıdır. Türkiye’de mevcut olan glukagon preparatları intramüsküler (IM), intravenöz (IV) ve subkutan (SC) kullanım için onaylı olsa da, üretici firma tarafından IV infüzyon şeklindeki kullanımı önerilmemektedir. Bu durumun temel sebebi, IV infüzyonla ilişkili olarak, steroid tedavisine direnç gösteren nekrotik migratuar eritem gibi ciddi komplikasyon riskinin bulunmasıdır. Tedaviye IM yolla başlanması durumunda önerilen doz 5-10 mg’dır; ancak bu uygulama yönteminde glukagonun emilimi daha yavaş olduğundan (5-30 dakika), klinik yanıtta gecikme veya yetersizlik olabilir ve tekrarlayan dozlara ihtiyaç duyulabilir. Bu nedenle, klinik durumun ciddiyeti göz önünde bulundurularak, tedaviye IV puşe şeklinde başlanması ya da IM uygulama sonrası yeterli klinik yanıt alınamadığında IV puşe ile devam edilmesi uygun olacaktır. Ancak IV infüzyon uygulamasından yukarıda belirtilen komplikasyon riski nedeniyle kaçınılması önemlidir. | |||
| İnsülin | Miyokardiyal disfonksiyon gelişen KKB veya Beta Blokör toksisitesi | 1 U/kg IV bolus, ardından: | 1-2 U/kg/h, her 15 dk’da bir titre edilir, maksimum infüzyon hızı 10 U/kg/h. |
| YAZAR NOTU: Ciddi KKB ve BB toksisitesinde HIET kullanımı en etkin tedavilerden biri olarak kabul edilmektedir ancak HIET tedavisine başlamadan önce hastanın klinik durumunun stabil olduğu dönemde yatak başı EKO ile hastanın bazal EF değeri hesaplanmalıdır. Hastanın klinik durumu bozulup HIET başlama endikasyonları doğduğu sırada EKO tekrarlanmalı ve EF’de düşme olup olmadığına bakılmalıdır zira HIET tedavisinin en etkili olduğu durum, BB ve KKB toksisitesine bağlı kardiyak EF’de bozulma durumudur. | |||
| Sodyum Bikarbonat | Geniş QRS’li disritmi (QRS>120 msn)Üriner alkalinizasyon | 1-2 mEq/kg %8,4 Sodyum bikarbonat IV bolus | 150 mEq/L sodyum bikarbonat, 1 litre %5 dekstroz içerisindeveya8.4% sodyum bikarbonat çözeltisi (1 mEq/mL) kullanılarak hazırlanabilir. |
| YAZAR NOTU: Sodyum kanal blokajına sebep olan ajanlara bağlı gelişen toksisitede blokajı çözmek için başlama sınırı QRS>100ms olmalıdır. Verilmesi gereken doz hesabı genellikle karışık olsa da pratik olarak hastanın kilosuna denk mEq dozunda sodyum bikarbonatı başlangıç bolus dozu olarak vermek en pratik yaklaşımdır. Örnek verecek olursak: 70 kg bir hastanın çekilen EKG’sinde sodyum kanal blokajı bulguları saptanan ve QRS 160 ms olan bir hastada bolus doz miktarı, 70 kg hasta için eşdeğer doz 70 mEq NaHCO3 olmalı, o da 7 ampul NaHCO3 eşdeğeri olup, hastaya bu doz uygulanmalı ve akabinde EKG çekilerek QRS’te daralma olup olmadığı izlenmelidir. QRS’te daralma yoksa doz tekrarlanmalı, daralma görüldüğü anda da infüzyon dozuna geçilmelidir. Hedef, QRS’te daralma olmasıdır, QRS’in normal aralığa dönmesi değildir. Yani QRS 160 ms iken 140 ms olmuşsa hedef etkiye ulaşılmış kabul edilir ve QRS < 100 ms olması beklenmeden infüzyon dozuna geçilir. İnfüzyon dozunun pratik hesabı da hastaya verilen IV bolus dozunun yarısıdır. Bu hasta örneğinde 7 ampul IV bolus yapıldıysa, infüzyon dozu bunun yarısı yani 3,5 ampul/saat dozunda olmalı ve en az 6 saat süreyle bu infüzyona devam edilmelidir. İnfüzyon bitiminde ise hastanın en az 24 saat monitörize biçimde ve aralıklı EKG çekilerek sodyum kanal blokajı bulguları açısından takip edilmesi gerekmektedir (referans A). | |||
| Digoksin Fab | Digoksin veya kardiyak glikozit zehirlenmesi durumundaHayatı tehdit edici disritmiHemodinamik unstabiliteHiperkalemi > 5 meq/L | Kardiyak arrestin yakın olduğu durumlarda başlangıçta ampirik olarak 400 mg (10 vial) uygulanabilir; diğer durumlarda ise 80 mg (2 vial) dozunda verilebilir. | Altta yatan kalp hastalığı nedeniyle kardiyak bozulma riski taşıyan hastalarda yarım molar geri dönüşüm düşünülebilir. |
| YAZAR NOTU: Digoksin zehirlenmelerine bağlı hayatı tehdit eden durumlarda DijiFab endike olsa da belirtilen dozları bulmak pek mümkün olmamaktadır. Zira DijiFab hem pahalı bir antidottur hem de gelişmiş ülkelerde bile kullanımı ciddi hastane maliyetlerine sebep olmaktadır. Bu yüzden son yıllarda DijiFab dozlarının modifiye edilmesi gündeme gelmiştir. İki yıl önce yayınlanan Avrupa kılavuzu ve iki ay önce yayınlanan Amerika kılavuzu bu sorunları gündeme almış ve DijiFab dozlarını güncellemiştir. Çevrilen bu makale 2020 yılında yayınlanmıştır. Bu sebeple yazar bu tabloda belirtilen dozları güncelleme ihtiyacı hissetmiştir. Yeni kılavuzlara göre kardiyak arrest durumunda serum digoksin düzeyini (SDD) beklemeden 10 değil ampirik olarak 5 vial DijiFab IV bolus dozu ile başlanıp 30 dk beklendikten sonra gerekirse doz tekrarı yapılması önerilmektedir. Kardiyak arrest olmaksızın hayatı tehdit eden ciddi zehirlenme durumlarında da yine SDD beklemeden ampirik olarak (hayatı tehdit eden aritmi, ciddi hipotansiyon vb.) 1-2 vial en az 30 dk olacak şekilde infüzyon (bolus değil) olarak verilip yine gereken durumda doz tekrarı yapılması önerilmektedir. Akut ve kronik digoksin toksisitesinde serum K seviyesine göre DijiFab başlama endikasyon eşik değeri hala net olarak bilinmemektedir, Kaynaklarda serum K seviyesi 5, 6 ve 6,5 olduğu durumlarda başlanması önerilse bile, şu an için SDD’yi beklemeden serum K>6 olduğu durumda DijiFab başlamak en mantıklı öneri olarak kabul edilebilir. Kardiyak arrest olmayan ve ciddi toksisite bulguları olmayan hastalarda ise önce SDD görülmesi ve buna göre kaç vial DijiFab verileceğine karar verilmesi önerilmektedir. Ayrıca son Avrupa kılavuzu SDD kullanılarak hesaplanan formülü de değiştirmiştir. Buna göre; yeni formül: SDD*hastanın kilosu (kg)*0,005=verilecek DijiFab vial sayısı. Bu formülden çıkan vial sayısının ise önce yarısının infüzyonla verilip, hastanın cevabına göre doz tekrarı denenebilir (referans C,D). | |||
| Lorazepam | İlaç ilişkili status epileptikus (first-line tedavi) | Nöbetler azalana kadar her 4-5 dk’da bir 4 mg IV | |
| YAZAR NOTU: Türkiye’de IV Lorazepam bulunmadığı için, ilaç ilişkili nöbetlerde IV Midazolam veya IV Diazepam başlanması önerilmektedir. Dozlar: Midazolam, 0.05–0,1 mg/kg IV, eğer IV yol sağlanamadıysa 0.1–0,2 mg/kg IM. Diazepam, 0.1–0,2 mg/kg IV. | |||
| Piridoksin | İzoniazid zehirlenmesiHidrazin zehirlenmesiGyromitra cinsi mantar zehirlenmeleriİnatçı ilaç ilişkili nöbet varlığı | İzoniazid alım miktarına eşdeğer gram cinsinden dozda uygulanmalıdırveyaerişkin hastalarda maksimum 5 g olacak şekilde, 25 mg/kg IV olarak 15-30 dk içinde verilebilir. | |
| Propofol | İlaç ilişkili status epileptikus | EEG’de burst supresyon sağlamak için propofol titrasyonu gereklidir (Genellikle>80 mcg/kg/dk). | |
| Naloksan | Opioid zehirlenmesi | Oksijenasyon ve ventilasyonun desteklendiği hastane ortamlarında, 0,04 mg IV dozunda her 1-2 dakikada bir titre edilerek solunum hızının >10/dk olması sağlanabilir; bu yaklaşım, ani yoksunluk sendromunu önleyebilir.IV erişim sağlanamadığında, 0.4–2 mg IM veya 4 mg intranazal olarak uygulanabilir.Başlangıç dozu etkisiz kalırsa tekrarlanabilir. Yeni veya yüksek potensli opioidler için >10 mg gibi daha yüksek dozlar gerekebilir. | Nalokson sonrası tekrarlayan solunum depresyonu veya yeniden sedasyon gelişen hastalarda, geri döndürme için gereken dozun 2/3’ü oranında IV infüzyona başlanmalıdır ve bu doz saatlik olarak uygulanmalıdır (örneğin,1 mg ile yanıt alındıysa, infüzyon 0,66 mg/h hızında başlatılmalıdır). |
| Hidroksikobalamin | Siyanür zehirlenmesi Refrakter vazopleji | 5 gr IV 15 dk içinde | 5 g doz, klinik duruma göre 15 dk ile 2 saat arasında IV infüzyon şeklinde tekrarlanabilir; toplam doz 10 g’a kadar çıkabilir. |
KARDİYOJENİK ŞOK ve VAZODİLATÖR ŞOK
Vazopressörler ve İnotroplar
Acil tıp hekimleri, hipotansiyon veya nedeni bilinmeyen şok durumundaki hastaların başlangıç resüsitasyonunda intravenöz sıvılar ve vazopressörleri yaygın olarak kullanmaktadır. Bu durum, ilaç kaynaklı şok gelişen hastaları da kapsamaktadır. Acil serviste POCUS’un yaygınlaşmasıyla birlikte, vazodilatör, kardiyojenik veya mikst şok ayrımı doğru şekilde yapılmaya başlanmıştır. Bu durum başlangıç resüsitasyonunda şokun türüne göre sıvı ihtiyacı/toleransı, vazopressör ve inotrop kullanımına yönelik ampirik tedavi yaklaşımını nasıl yönetileceğine dair klinisyene fikir vermektedir. Örneğin, dihidropiridin türevi kalsiyum kanal blokörleri başlangıçta vazodilatasyona bağlı şoka neden olabilir ve yalnızca kalsiyum ve vazopressör tedavisine yanıt verebilir. Ancak, non-dihidropiridin türevi diğer kalsiyum kanal blokörleri veya yüksek dozda alındığında reseptör seçiciliğini kaybeden dihidropiridinler, miyokard depresyonu ile seyreden kardiyojenik şoka yol açabilir ve bu durumda inotrop ajanların kullanımı daha uygun olabilir.
YAZAR NOTU: Dihidropiridinler (örn. amlodipin, nifedipin gibi kalsiyum kanal blokerleri) normal dozlardaL tipi kalsiyum kanallarına spesifik olarak bağlanır. Ancakçok yüksek dozlarda alındığında, bu ilaçlarınreseptör seçiciliği kaybolabilir ve başka iyon kanallarına veya dokulara da etki edebilir. Bu sebeple Dihidropiridinler düşük dozda sadecevasküler düz kasları gevşeterek hipotansiyon oluşturur ancak yüksek dozda kardiyak ve santral sinir sistemi üzerindeki etkileri ortaya çıkabilir.
İlaç kaynaklı şok tedavisinde vazopressör kullanımına ilişkin endişeler, ağırlıklı olarak hayvan modellerine dayanan iskemik komplikasyonlar, kardiyak metabolizma üzerindeki olumsuz etkiler ve kardiyak debi üzerindeki advers etkiler ile ilişkilidir. Ancak Verapamil ve diltiazem doz aşımı nedeniyle yüksek doz vazopressör ve inotrop tedavisi uygulanan hastaların değerlendirildiği tek merkezli retrospektif bir çalışmada, iskemi komplikasyonları düşük, sağkalım oranları ise yüksek bulunmuştur. Bir başka sistematik derlemede de toksinlerin sebep olduğu kardiyojenik şok tedavisinde vazopressörlerin kullanımının belirgin zararlı etkilerinin bulunmadığı ve hastaların büyük çoğunluğunda sağkalım oranlarının yüksek olduğu bildirilmiştir. Bununla birlikte, araştırmacılar bu derlemede vazopressör tedavisinin başarısızlıklarının yeterince raporlanmadığını belirtirken, hipotansiyonun vazopressörlerle düzeltilmesinin bildirilmesi veya vaka takdimi olarak hazırlanması gereken önemli bir tıbbi durum olarak görülmemesi sebebiyle, vazopressör kullanımına dair tedavi başarılarının da eksik bildirilme olasılığının bulunduğunu vurgulamaktadır. Kalsiyum kanal blokeri zehirlenmelerinin tedavisine yönelik yapılan bir sistematik incelemede, dopamin ve norepinefrinin hemodinamik parametreleri ve sağkalımı ciddi yan etki olmadan iyileştirdiği, ancak mevcut kanıt kalitesinin oldukça düşük olduğu rapor edilmiştir. Bu nedenle, sıvı volüm durumu optimize edildikten sonra, kardiyovasküler ilaç toksisitesi gelişen hastalarda hemodinamik parametreleri desteklemek amacıyla vazopressör veya inotrop ajanların uygulanması makul bir yaklaşımdır. Bu süreç, ideal olarak invaziv (pulmoner arter kateterizasyonu) veya non-invaziv (ekokardiyografi, nabız kontur analizi) hemodinamik monitorizasyon ile yönetilmelidir.
Hiperinsülinemik Öglisemik Tedavi (HIET)
Hiperinsülinemik-öglisemik tedavi (HIET), öncelikle kalsiyum kanal blokeri ve beta bloker zehirlenmelerinde kullanılan bir tedavi yöntemi olup, genellikle öglisemiyi sağlamak amacıyla konsantre dekstroz infüzyonları ile kombine edilen çok yüksek dozlarda insülin uygulanmasını içerir. İlaç toksisitesine bağlı olarak fonksiyon kaybına uğramış kardiyak miyositler, enerji metabolizmalarını öncelikli olarak kullandığı serbest yağ asitlerinden karbonhidrat metabolizmasına yönlendirir. Hayvan modellerinde yapılan çalışmalarda, ilaç kaynaklı kardiyojenik şok durumunda insülin uygulamasının hem sistolik hem de diyastolik kardiyak fonksiyonları iyileştirdiği ve yetmezlik gelişmiş insan miyokardı üzerinde bağımsız pozitif inotropik etkiler gösterdiği bildirilmiştir. Kalsiyum kanal blokeri zehirlenmesi insülin direncine yol açmaktadır, ayrıca pankreastaki L-tipi kalsiyum kanallarının antagonizmasına yol açarak pankreastan kalsiyum aracılığıyla insülin salınımını da inhibe etmektedir. Bu etkiler, fizyolojik stres yanıtı ile birleştiğinde hiperglisemi ve relatif hipoinsülinemi gelişimine neden olmaktadır. Nitekim, verapamil ve diltiazem doz aşımını içeren retrospektif bir çalışmada, hiperglisemi derecesinin hastalığın şiddetini belirlemede hemodinamik parametrelerden daha iyi bir gösterge olduğu bildirilmiştir. HIET’in antidotal mekanizması, değişen kardiyak metabolizma ihtiyaçlarını karşılamak, inotropiyi artırmak ve periferik vazodilatasyonu destekleyerek organ perfüzyonunu iyileştirmek üzerine kuruludur. Bu nedenle, HIET antidotunun en iyi kullanım alanı, miyokard kontraktilitesinin bozulduğu beta bloker veya kalsiyum kanal blokerlerinin sebep olduğu kardiyojenik şok olgularının tedavisidir.
Kalsiyum kanal blokeri zehirlenmelerinin tedavisine yönelik güncel kılavuzlar, HIET’i birinci basamak tedaviler arasında önermektedir. Tedavi başlatıldıktan sonra, insülin infüzyon hızları klinik yanıta göre titre edilebilir. Öglisemiyi sağlamak için genellikle 25 gr/saat dozunda %50 dekstroz infüzyonu uygulanmaktadır. Tüm infüzyonların, ilaca bağlı etkiler, volüm yüklenmesi veya akut sol ventrikül disfonksiyonu nedeniyle gelişebilecek pulmoner ödemi önlemek amacıyla konsantre edilmesi önerilmektedir. Hipokalemi, hiponatremi ve hipoglisemi, zehir danışma merkezleri veya toksikolog gözetiminde dahi sık görülebilen yan etkilerdir ve tedavi sürecinde dikkatle izlenmelidir.
Glukagon
Pankreatik hormon glukagon, G-protein aracılıklı glukagon reseptörünü aktive ederek β₁-adrenerjik reseptörün toksik etkisini dolaylı yoldan bloke eder. Bu etki, adenilat siklazın uyarılması ve hücre içi siklik AMP seviyesinin artmasıyla sonuçlanır. Bu mekanizma, hayvan modellerinde beta bloker zehirlenmesinde farmakolojik olarak artmış inotropi ve kronotropi ile sonuçlanır. Ancak, insanlarda beta bloker doz aşımında glukagon kullanımına ilişkin sonuçlar çelişkili bulunmuştur. Propranolol ve verapamil toksisitesine yönelik hayvan modellerinde, glukagonun antidotal etkinliğinin HIET’e kıyasla daha düşük olduğu bulunmuştur. Bununla birlikte, glukagonun yan etki profili genellikle iyi tolere edilir ve doz bağımlı olarak bulantı, kusma ve hiperglisemi görülebilir. Bilinci korunan, hava yolu refleksleri yeterli olan veya entübe edilmiş hastalarda, miyokardiyal disfonksiyon veya kardiyojenik şok ile seyreden beta bloker toksisitesinde glukagon kullanımının uygun bir seçenek olabileceği bildirilmiştir. Eğer bolus doz uygulamasına olumlu klinik yanıt alınırsa, yanıtın sağlandığı miligram cinsinden dozun saatlik infüzyon şeklinde devam ettirilmesi önerilmektedir.
Metilen Mavisi
Metilen mavisi, nitrik oksit üretim yolaklarındaki çözünür guanilat siklaz ve nitrik oksit sentazı inhibe eder. Bu inhibisyon mekanizması, vazodilatasyonun azalmasına ve sistemik vasküler direncin artmasına neden olur. Bu nedenle Metilen mavisinin refrakter vazodilatör şok tedavisinde kullanılması önerilmektedir. İlaç kaynaklı şok tedavisinde metilen mavisinin kullanım etkinliğini değerlendiren yalnızca olgu sunumları ve özetlerin bulunduğu sistematik bir derlemede, hemodinamik etkilerinin değişkenlik gösterdiği bildirilmiştir. Verilerin randomizasyondan yoksun olması, yayın yanlılığı, diğer tedavilerle etkileşim ve eksik raporlama gibi sınırlamalar nedeniyle, araştırmacılar metilen mavisinin ilaç kaynaklı şok tedavisinde rutin olarak önerilebilmesi için yetersiz kanıt bulunduğunu belirtmiştir. Metilen mavisi, hemoliz riski nedeniyle glukoz-6-fosfat dehidrojenaz (G6PD) eksikliği olan hastalarda kontrendikedir. Daha önce incelenen vakalarda ciddi advers etkiler bildirilmemiş olmakla birlikte, serotonerjik ilaç kullanan hastalarda serotonin sendromunu tetikleyebileceği de bilinmelidir.
Hidroksikobalamin
Hidroksikobalamin, siyanür antidotu olarak iyi bilinen ve etkinliği kanıtlanmış bir tedavi yöntemidir. Bilinç kaybı, nöbet ve kardiyak veya solunumsal yetmezlik gibi ağır siyanür maruziyeti durumlarında, hidroksikobalamin ile ampirik tedavinin uygulanmasının belirgin bir sakıncası bulunmamaktadır. Acil serviste siyanür zehirlenmesinin kanıta dayalı tedavi algoritmalarından biri, duman inhalasyonu sonrası olası siyanür zehirlenmesi düşünülen hastalarda, şiddetli maruziyet düşünülüyorsa, hidroksikobalamin ile acil ampirik tedavi önermektedir.
Hidroksikobalamin ayrıca, nitrik oksidi (NO) bağlayarak NO aracılıklı vazoplejiyi tersine çevirme yeteneğine dayanarak refrakter vazodilatör şok için kurtarma tedavisi olarak kullanılmıştır. Bu etki, ilaçla yapılan gönüllü çalışmalar sırasında hipertansif yanıt olarak gözlemlenmiş ve daha sonra kardiyak cerrahide, kardiyopulmoner bypass sonrası gelişen vazoplejik sendromun tedavisinde başarılı bir şekilde kullanılmıştır. Ancak, siyanür dışı doz aşımı vakalarından kaynaklı şokun tedavisinde, hidroksikobalamin kullanımına ilişkin herhangi bir rapor bulunmamaktadır. Hidroksikobalaminin normal dozunun 5 katın üzerindeki iatrojenik aşırı doz uygulanan bir vakada, gözlenen tek klinik belirti kendiliğinden düzelen eritrodermaolmuştur. Siyanür zehirlenmesi tedavisinde kullanılan standart dozda uygulandığında, hidroksikobalaminin yan etki profili oldukça iyi tolere edilebilir olup, sınırlı kanıt bulunmasına rağmen ilaç kaynaklı refrakter vazoplejinin tedavisinde son basamak olarak kullanımı değerlendirilebilir.
Toksikoloji Hastalarında Mekanik Dolaşım Desteği
Kritik derecede zehirlenmiş hastalar, ekstrakorporeal yaşam desteği (ECLS) için ideal adaylar olabilir. Doz aşımı vakalarındaki hastalar genellikle daha genç hastalar olup, kardiyak endikasyonlarla ECLS yapılması değerlendirilen hastalara kıyasla daha az komorbiditeye sahiptir. Kritik derecede zehirlenmiş bu hastalar, toksisitenin ortadan kalkmasına kadar desteklenebilirse, tam iyileşme olasılığı oldukça yüksektir. Ancak buna rağmen, zehirlenmiş hastalarda ECLS kullanımı nadir olarak bildirilmektedir. Ayrıca intraaortik balon pompası ve Impella (Abiomed, Danvers, MA, USA) perkütan sol ventrikül destek cihazı gibi mekanik dolaşım destek cihazlarının zehirlenmiş hastalarda kullanıldığı rapor edilse de bu cihazlara ilişkin kanıtlar da oldukça sınırlıdır.
Her ne kadar yaygın uygulanmasa da ECMO, zehirlenmiş hastalar için en sık bildirilen mekanik destek yöntemi olmuştur. Literatürde acil perkütan ECMO uygulanan hastaları içeren bir seride, zehirlenmiş hastalardan elde edilen sonuçlar, primer kardiyak endikasyonlarla ECMO uygulanan hastalara kıyasla daha başarılı bulunmuştur. Başka bir çalışmada, zehirlenmeye bağlı şok nedeniyle ECMO uygulanan 12 hasta ile kardiyovasküler nedenlerle ECMO uygulanan 5 hasta karşılaştırılmıştır. Kanülasyon sırasında tüm hastaların 45 dakikadan uzun süre devam eden kardiyopulmoner resüsitasyona (CPR) ihtiyaç duyduğubelirtilmiştir. ECMO uygulanan zehirlenmiş hastaların 3’ü sağ kalırken, kardiyovasküler endikasyon grubundaki hiçbir hasta hayatta kalamamıştır. Masson ve çalışma arkadaşları, persistan şok veya kardiyak arrest gelişen 62 zehirlenmiş hastayı incelemiş ve bu hastaların 14’ü venö-arteriyel (VA) ECMO ile tedavi edilmiştir. ECMO uygulanan grupta sağkalım oranı %86 iken, konvansiyonel yöntemlerle tedavi edilen grupta sağkalım %48 olarak bildirilmiştir. Ancak, çalışmadaki hasta sayısının sınırlı olması nedeniyle sonuçların tartışmaya açık olduğu belirtilmiştir. Extracorporeal Life Support Organization (ELSO) veritabanında yapılan bir analizde, zehirlenmiş hastalar için ECMO kullanımının giderek arttığı ve genel sağkalım oranının %59 olduğu bildirilmiştir. İnhalasyon veya aspirasyon hasarı nedeniyle venö-venöz (V-V) ECMO uygulanan hastalarda ise sağkalım oranı %89 olarak tespit edilmiştir. Konvansiyonel tedaviye yanıt vermeyen ağır refrakter hipoksi, persistan kardiyojenik şok veya kardiyak arrest gelişen zehirlenmiş hastalar, ECMO’nun mevcut olduğu kurumlarda ECMO açısından değerlendirilmelidir. ECMO uygulama kapasitesine sahip olmayan merkezlerde bir ECMO transport ekibi bulunuyorsa, konvansiyonel tedaviyle yüksek mortalite riski taşıyan bu kritik hastalar için acil olarak ECMO transport ekibiyle konsültasyon sağlanmalıdır, çünkü bu hasta grubunda ECLS sağkalım oranları oldukça yüksektir.
İlaç İlişkili Karaciğer Yetmezliği
Akut fulminan hepatik yetmezlik olarak da adlandırılan akut karaciğer yetmezliği (ALF), altta yatan bir karaciğer hastalığı olmayan bir hastada, akut (< 26 hafta) bir hasar sonucu gelişen hepatik hasar, ensefalopati ve sentetik fonksiyon bozukluğu (INR > 1.5) olarak tanımlanır. ABD ve Avrupa’da ALF’nin en yaygın nedeni ilaçlardır. ABD Akut Karaciğer Yetmezliği Çalışma Grubu (US ALF Study Group) kayıtlarına göre, vakaların çoğunu asetaminofen (APAP) ilişkili ALF oluştururken, asetaminofen dışı nedenlere bağlı ALF vakaları toplam olguların %11’ini oluşturmaktadır. Asetaminofen ilişkili ALF gelişen hastaların, hastalığın ileri evresinde başvurma eğiliminde olduğu bildirilmiştir zira asetaminofenin toksik metaboliti olan N-asetil-p-benzokinonimin (NAPQI) aracılığıyla karaciğer hasarının oluşması için belirli bir zaman gerekmektedir. ALF ile başvuran hastalarda belirgin transaminaz yüksekliği, düşük bilirubin seviyesi ve yüksek INR değerleri, APAP ilişkili ALF’yi düşündüren en önemli laboratuvar bulgularıdır.
N-asetilsistein (NAC), hepatik glutatyon depolarını yenileyerek, NAPQI’nin detoksifikasyonunu sağlayarak ve antiinflamatuar özellik göstererek, APAP doz aşımına bağlı ALF’de sağkalımı artırdığı gösterilmiş bir tedavidir. NAC, doğru dozda uygulandığında güvenli bir yan etki profiline sahiptir. ALF gelişen tüm erişkin hastalara, APAP düzeyi saptanamaz hale gelene ve aminotransferazlar ile klinik biyobelirteçler (kreatinin, laktat, pH, protrombin zamanı/INR, fosfat) iyileşene kadar, asetaminofen antidot protokolüne uygun olarak NAC tedavisi verilmelidir. Bazı olgularda NAC bolus uygulaması veya daha yüksek infüzyon hızlarında verilmesi gerekebilir ve bu süreç toksikoloji uzmanı ile değerlendirilmelidir. Koagülopatinin düzeltilmesi genellikle önerilmez, ancak klinik olarak anlamlı bir kanama varlığında veya invaziv prosedürler için gerekiyorsa uygulanabilir. Çünkü koagülopati derecesi, karaciğer nakli gereksinimini öngören tüm prognostik modelleri etkileyen önemli bir parametredir.
Hiperamonyemi, astrositlerde glutamine dönüşüm yoluyla serebral ödem ve intrakraniyal basınç (ICP) artışına neden olabilir. Ancak, ALF’de amonyak düzeylerini düşürmek için laktüloz veya rifaksimin kullanımına dair kanıt bulunmamaktadır. Refrakter hiperamonyemi (>100 µmol/L) veya intrakraniyal hipertansiyon riski yüksek olan ciddi ensefalopati durumlarında, sürekli renal replasman tedavisi (CRRT) ile amonyak seviyeleri düşürülebilir, ancak bu tedaviye ilişkin kanıtlar sınırlıdır. ALF hastalarında invaziv ICP monitorizasyonunun risk ve yararları konusunda kabul edilmiş bir görüş birliği bulunmamaktadır ve uygulamalar kurumdan kuruma farklılık göstermektedir.
ALF için sağkalımı kanıtlanmış tek diğer tedavi acil karaciğer naklidir, ancak elektif karaciğer nakli geçiren hastalara kıyasla sağkalım oranları daha düşüktür. Hem APAP hem de diğer tüm nedenlere bağlı gelişen ALF’nin prognozunu değerlendirmek için çeşitli kriterler geliştirilmiştir. En yaygın kullanılanlardan ikisi King’s College Kriterleri (APAP ve APAP dışı nedenler için ayrı setler içermektedir) ve Son Dönem Karaciğer Hastalığı Modeli (MELD) skorudur. King’s College Kriterleri daha spesifik, MELD skoru ise ilaç kaynaklı ALF’de karaciğer nakli ihtiyacını belirleme açısından daha sensitif bulunmuştur. İlaç ilişkili ALF şüphesi olan hastalar için, acil hepatoloji konsültasyonu yapılmalı ve mümkünse karaciğer nakli merkezine transfer edilmelidir.
Eliminasyonu artırıcı tedavi
Pek çok ilacın eliminasyonu idrarın alkali hale getirilmesiyle artırılabilse de klinik olarak idrar alkalinizasyonunun en etkin olduğu ilaçlar salisilat, metotreksat ve fenobarbitaldir. Sodyum bikarbonat kullanılarak idrar pH’ının 8 civarında tutulması, bu toksinlerin renal eliminasyonunu artırmaktadır. Salisilat zehirlenmesi durumunda hem kanın hem de idrarın alkali hale getirilmesi, yalnızca eliminasyonu artırmakla kalmaz, aynı zamanda salisilatın dağılım hacmini azaltarak merkezi sinir sistemi üzerindeki toksik etkilerini de en aza indirmeye yardımcı olur.
Ekstrakorporeal Toksin Eliminasyonu (ECTR)
Kritik toksikoloji hastalarının birçoğunda, metabolik asidoz, elektrolit dengesizlikleri, volüm yüklenmesi veya üremi gibi klasik hemodiyaliz endikasyonlarıyla seyreden akut böbrek hasarı gelişebilir. Bu durumlarda hastanın nefroloji ekibine erken dönemde danışılması kritik olup, toksin eliminasyonu ikincil bir öneme sahiptir. Ekstrakorporeal toksin eliminasyonu (ECTR), hemodiyaliz veya sürekli renal replasman tedavisi (CRRT) ile sağlanabilir. Teknik ilerlemeler ve toksikokinetik süreçlerin daha iyi anlaşılması, klasik ECTR kriterlerini değiştirmiştir. Yüksek etkinlikli, yüksek akışlı diyaliz membranları, 15.000 Da’ya kadar olan maddelerin eliminasyonunu artırırken, daha geniş çaplı hemodiyaliz kateterleri ve gelişmiş hemodiyaliz cihazları, diyaliz sırasında daha yüksek kan akış hızlarına olanak tanımaktadır. Proteinlere bağlanma oranı yüksek olan salisilat, valproik asit, fenitoin ve karbamazepin gibi bazı ilaçlar, aşırı doz durumunda ECTR’ye uygun hale gelebilir. Bu ilaçlarda, protein bağlanma kapasitesi doygunluğa ulaştığında serbest ilaç fraksiyonu artar ve hemodiyaliz ile serbest halde olan ilaç fraksiyonu uzaklaştırılabilir.
İlaçların aşırı doz olması durumunda ECTR’nin etkili bir tedavi seçeneği haline gelebilmesi için; bir toksinin ekstrakorporeal klirensinin toplam vücut klirensinin önemli bir kısmını oluşturması gereklidir. Ayrıca, toksine karşı etkili bir antidot bulunmamalıdır; aksi takdirde, antidot tedavisinin risk/fayda oranı ECTR’yi gereksiz kılabilir. Örneğin, insülinin kendisi yüksek akışlı membranlar ile diyaliz edilebilir, ancak hipoglisemi durumunda yoğun dekstroz tedavisi kolaylıkla uygulanabildiğinden diyaliz gibi ECTR tedavileri genellikle gereksizdir.
CRRT, hemodinamik olarak stabil olmayan yoğun bakım hastalarında sıklıkla tercih edilmekle birlikte, bu yöntemle ilaç ve toksin eliminasyonu genellikle yetersiz düzeydedir. Bununla birlikte, CRRT; akut karaciğer yetmezliği olan hastalarda net sıvı uzaklaştırmasını sağlamak ve serebral perfüzyon basıncını korumak açısından faydalı olabilir. Genel olarak, ECTR endikasyonu tek bir ilaç düzeyine dayanarak belirlenmemelidir; zira kan düzeyi genellikle hedef organdaki toksisiteyi tam olarak yansıtmaz. Bunun yerine, ciddi organ disfonksiyonu gelişen veya ECTR ile uzaklaştırılabilen, yaşamı tehdit eden toksinlere maruz kalan hastalarda, acil nefroloji konsültasyonu ile hemodiyaliz uygulanması düşünülmelidir.
ÖZET
Kritik toksikoloji hastaları, acil tıp hekimleri için benzersiz kritik bakım gereksinimleri sunar. Bu hastalar sıklıkla hayati tehlike arz eden bir klinik durumda acil servise başvurur ve zehirlenmeye bağlı gelişen klinik durumları için sıklıkla spesifik antidot ve tedavi ihtiyaçları bulunur. Acil tıp hekimlerinin aşina olduğu bu tablolar arasında status epileptikus, kardiyojenik şok, akut böbrek hasarı ve karaciğer yetmezliği gibi durumlar yer alır. Zehirlenmeye bağlı kritik durumdaki hastaların resüsitasyonu büyük ölçüde acil servis sorumluluğundadır; ancak kritik durumdaki bu hastaların tamamı sonuçta yoğun bakım ünitesi yönetimi gerektirir. Bununla birlikte, mekanik dolaşım desteği ihtiyacının acil olarak değerlendirilmesi, organ nakli için uygunluk değerlendirmesi ve ekstrakorporeal toksin eliminasyonu veya renal replasman tedavisi gibi kritik kararlar genellikle acil serviste, acil tıp hekimleri tarafından verilmelidir. Bu nedenle, hızlı tanısal değerlendirme, hemodinamik stabilizasyon ve spesifik ilaç veya ilaç grubu antidotlarının uygulanması hasta sağkalımını ve klinik sonuçları iyileştirmek açısından kritik öneme sahiptir.
Yazar Referansları
Referans A
Boehnert MT, Lovejoy Jr FH. Trisiklik antidepresanların akut aşırı dozundan sonra nöbetleri ve ventriküler aritmileri tahmin etmede QRS süresinin serum ilaç düzeyine göre değeri. N Engl J Med. 1985;313(8):474–9. doi: 10.1056/nejm198508223130804 .
Referans B
Nakatsu, L., Lopez, J. R., Garcia, C. M., Cherian, M., Nash, J., Tofighi, D., … & Warrick, B. J. (2025). Comparison of two-bag and three-bag acetylcysteine regimens in the treatment of paracetamol poisoning: a systematic review and meta-analysis. Clinical Toxicology, 1-11.
Referans C
Hack, J. B., Wingate, S., Zolty, R., Rich, M. W., & Hauptman, P. J. (2024). Expert Consensus on the Diagnosis and Management of Digoxin Toxicity. The American Journal of Medicine.
Referans D
Andrews, P., Anseeuw, K., Kotecha, D., Lapostolle, F., & Thanacoody, R. (2023). Diagnosis and practical management of digoxin toxicity: a narrative review and consensus. European Journal of Emergency Medicine, 30(6), 395-401.
Makale Referansları
1. Mazer-Amirshahi M, Sun C, Mullins P, et al. Trends in emergency department resource utilization for poisoning-related visits, 2003-2011. J Med Toxicol 2016; 12(3):248–54.
2. Prevention CfDCa. Available at: https://www.cdc.gov/nchs/fastats/injury.htm. Accessed November 28, 2019.
3. Chen HY, Albertson TE, Olson KR. Treatment of drug-induced seizures. Br J Clin Pharmacol 2016;81(3):412–9.
4. Hocker SE, Britton JW, Mandrekar JN, et al. Predictors of outcome in refractory status epilepticus. JAMA Neurol 2013;70(1):72–7.
5. Shah AS, Eddleston M. Should phenytoin or barbiturates be used as second-line anticonvulsant therapy for toxicological seizures? Clin Toxicol (Phila) 2010;48(8): 800–5.
6. Silbergleit R, Durkalski V, Lowenstein D, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med 2012;366(7):591–600.
7. Lheureux P, Penaloza A, Gris M. Pyridoxine in clinical toxicology: a review. Eur J Emerg Med 2005;12(2):78–85.
8. Rossetti AO, Reichhart MD, Schaller MD, et al. Propofol treatment of refractory status epilepticus: a study of 31 episodes. Epilepsia 2004;45(7):757–63.
9. Rosati A, De Masi S, Guerrini R. Ketamine for refractory status epilepticus: a systematic review. CNS Drugs 2018;32(11):997–1009.
10. Schmidt S, Schmitz-Buhl M. Signs and symptoms of carbamazepine overdose. J Neurol 1995;242(3):169–73.
11. DeLorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 1998; 39(8):833–40.
12. Beauchamp GA, Hendrickson RG, Hatten BW. Endotracheal intubation for toxicologic exposures: a retrospective Review of toxicology investigators consortium (ToxIC) cases. J Emerg Med 2016;51(4):382–8.e1.
13. van Dorp E, Yassen A, Dahan A. Naloxone treatment in opioid addiction: the risks and benefits. Expert Opin Drug Saf 2007;6(2):125–32.
14. Farkas A, Lynch MJ, Westover R, et al. Pulmonary complications of opioid overdose treated with naloxone. Ann Emerg Med 2020;75(1):39–48.
15. Somerville NJ, O’Donnell J, Gladden RM, et al. Characteristics of fentanyl overdose- Massachusetts, 2014-2016. MMWR Morb Mortal Wkly Rep 2017;66(14):382–6.
16. Otani Y, Kanno K, Toh Yoon EW, et al. Acute respiratory distress syndrome caused by salicylate intoxication. Clin Case Rep 2018;6(9):1905–6.
17. Ramanathan K, Tan CS, Rycus P, et al. Extracorporeal membrane oxygenation for poisoning in adult patients: outcomes and predictors of mortality. Intensive Care Med 2017;43(10):1538–9.
18. Levine M, Brooks DE, Truitt CA, et al. Toxicology in the ICU: Part 1: general overview and approach to treatment. Chest 2011;140(3):795–806.
19. Bass M. Sudden sniffing death. JAMA 1970;212(12):2075–9.
20. Al-Abri SA, Woodburn C, Olson KR, et al. Ventricular dysrhythmias associated with poisoning and drug overdose: a 10 year review of statewide poison control center data from California. Am J Cardiovasc Drugs 2015;15(1):43–50.
21. Levine M, Boyer EW, Pozner CN, et al. Assessment of hyperglycemia after calcium channel blocker overdoses involving diltiazem or verapamil. Crit Care Med 2007;35(9):2071–5.
22. Delk C, Holstege CP, Brady WJ. Electrocardiographic abnormalities associated with poisoning. Am J Emerg Med 2007;25(6):672–87.
23. Kerns W 2nd. Management of beta-adrenergic blocker and calcium channel antagonist toxicity. Emerg Med Clin North Am 2007;25(2):309–31 [abstract: viii].
24. Chan BS, Buckley NA. Digoxin-specific antibody fragments in the treatment of digoxin toxicity. Clin Toxicol (Phila) 2014;52(8):824–36.
25. Seger DL, Hantsch C, Zavoral T, et al. Variability of recommendations for serum alkalinization in tricyclic antidepressant overdose: a survey of U.S. Poison Center medical directors. J Toxicol Clin Toxicol 2003;41(4):331–8.
26. Bruccoleri RE, Burns MM. A literature review of the use of sodium bicarbonate for the treatment of QRS widening. J Med Toxicol 2016;12(1):121–9.
27. Litz RJ, Popp M, Stehr SN, et al. Successful resuscitation of a patient with ropivacaine-induced asystole after axillary plexus block using lipid infusion. Anaesthesia 2006;61(8):800–1.
28. Levine M, Hoffman RS, Lavergne V, et al. Systematic review of the effect of intravenous lipid emulsion therapy for non-local anesthetics toxicity. Clin Toxicol (Phila) 2016;54(3):194–221.
29. Hayes BD, Gosselin S, Calello DP, et al. Systematic review of clinical adverse events reported after acute intravenous lipid emulsion administration. Clin Toxicol (Phila) 2016;54(5):365–404.
30. Lee HM, Archer JR, Dargan PI, et al. What are the adverse effects associated with the combined use of intravenous lipid emulsion and extracorporeal membrane oxygenation in the poisoned patient? Clin Toxicol (Phila) 2015;53(3):145–50.
31. Gosselin S, Hoegberg LC, Hoffman RS, et al. Evidence-based recommendations on the use of intravenous lipid emulsion therapy in poisoning. Clin Toxicol (Phila) 2016;54(10):899–923.
32. Skoog CA, Engebretsen KM. Are vasopressors useful in toxin-induced cardiogenic shock? Clin Toxicol (Phila) 2017;55(4):285–304.
33. Levine M, Curry SC, Padilla-Jones A, et al. Critical care management of verapamil and diltiazem overdose with a focus on vasopressors: a 25-year experience at a single center. Ann Emerg Med 2013;62(3):252–8.
34. St-Onge M, Dube PA, Gosselin S, et al. Treatment for calcium channel blocker poisoning: a systematic review. Clin Toxicol (Phila) 2014;52(9):926–44.
35. Kline JA, Leonova E, Raymond RM. Beneficial myocardial metabolic effects of insulin during verapamil toxicity in the anesthetized canine. Crit Care Med 1995; 23(7):1251–63.
36. Kline JA, Raymond RM, Leonova ED, et al. Insulin improves heart function and metabolism during non-ischemic cardiogenic shock in awake canines. Cardiovasc Res 1997;34(2):289–98.
37. von Lewinski D, Bruns S, Walther S, et al. Insulin causes [Ca21]i-dependent and [Ca21]i-independent positive inotropic effects in failing human myocardium. Circulation 2005;111(20):2588–95.
38. Kline JA, Raymond RM, Schroeder JD, et al. The diabetogenic effects of acute verapamil poisoning. Toxicol Appl Pharmacol 1997;145(2):357–62.
39. St-Onge M, Anseeuw K, Cantrell FL, et al. Experts consensus recommendations for the management of calcium channel blocker poisoning in adults. Crit Care Med 2017;45(3):e306–15.
40. Cole JB, Arens AM, Laes JR, et al. High dose insulin for beta-blocker and calcium channel-blocker poisoning. Am J Emerg Med 2018;36(10):1817–24.
41. Krenz JR, Kaakeh Y. An overview of hyperinsulinemic-euglycemic therapy in calcium channel blocker and beta-blocker overdose. Pharmacotherapy 2018; 38(11):1130–42.
42. Beavers JR, Stollings JL, Rice TW. Hyponatremia induced by hyperinsulinemiaeuglycemia therapy. Am J Health Syst Pharm 2017;74(14):1062–6.
43. Love JN, Leasure JA, Mundt DJ, et al. A comparison of amrinone and glucagon therapy for cardiovascular depression associated with propranolol toxicity in a canine model. J Toxicol Clin Toxicol 1992;30(3):399–412.
44. Kerns W 2nd, Schroeder D, Williams C, et al. Insulin improves survival in a canine model of acute beta-blocker toxicity. Ann Emerg Med 1997;29(6):748–57.
45. Mayer B, Brunner F, Schmidt K. Inhibition of nitric oxide synthesis by methylene blue. Biochem Pharmacol 1993;45(2):367–74.
46. Warrick BJ, Tataru AP, Smolinske S. A systematic analysis of methylene blue for drug-induced shock. Clin Toxicol (Phila) 2016;54(7):547–55.
47. SAS P. Prescribing Information. 2016. Available at: https://www.americanregent.com/media/1802/provayblue-prescribing-information.pdf. Accessed November 27, 2019.
48. Lo JC, Darracq MA, Clark RF. A review of methylene blue treatment for cardiovascular collapse. J Emerg Med 2014;46(5):670–9.
49. Hamad E, Babu K, Bebarta VS. Case files of the university of massachusetts toxicology fellowship: does this smoke inhalation victim require treatment with cyanide antidote? J Med Toxicol 2016;12(2):192–8.
50. Jentzer JC, Vallabhajosyula S, Khanna AK, et al. Management of refractory vasodilatory shock. Chest 2018;154(2):416–26.
51. Burnes ML, Boettcher BT, Woehlck HJ, et al. Hydroxocobalamin as a rescue treatment for refractory vasoplegic syndrome after prolonged cardiopulmonary bypass. J Cardiothorac Vasc Anesth 2017;31(3):1012–4.
52. Friedman BT, Chen BC, Latimer AJ, et al. Iatrogenic pediatric hydroxocobalamin overdose. Am J Emerg Med 2019;37(7):1394.e1-2.
53. SAS MS. Cyanokit administration guide. Available at: https://www.cyanokit.com/ sites/default/files/CYANOKIT_Administration_Guide_PP-CYA-USA-0091.pdf. Accessed January 29, 2020.
54. Wang GS, Levitan R, Wiegand TJ, et al. Extracorporeal membrane oxygenation (ECMO) for severe toxicological exposures: review of the toxicology investigators consortium (ToxIC). J Med Toxicol 2016;12(1):95–9.
55. Vanzetto G, Akret C, Bach V, et al. [Percutaneous extracorporeal life support in acute severe hemodynamic collapses: single centre experience in 100 consecutive patients]. Can J Cardiol 2009;25(6):e179–86.
56. Megarbane B, Leprince P, Deye N, et al. Emergency feasibility in medical intensive care unit of extracorporeal life support for refractory cardiac arrest. Intensive Care Med 2007;33(5):758–64.
57. Masson R, Colas V, Parienti JJ, et al. A comparison of survival with and without extracorporeal life support treatment for severe poisoning due to drug intoxication. Resuscitation 2012;83(11):1413–7.
58. Khan R, Koppe S. Modern management of acute liver failure. Gastroenterol Clin North Am 2018;47(2):313–26.
59. Thomas AM, Lewis JH. Nonacetaminophen drug-induced acute liver failure. Clin Liver Dis 2018;22(2):301–24.
60. McPhail MJ, Kriese S, Heneghan MA. Current management of acute liver failure. Curr Opin Gastroenterol 2015;31(3):209–14.
61. ACMT position statement: duration of intravenous acetylcysteine therapy following acetaminophen overdose. J Med Toxicol 2017;13(1):126–7.
62. Rajajee V, Fontana RJ, Courey AJ, et al. Protocol based invasive intracranial pressure monitoring in acute liver failure: feasibility, safety and impact on management. Crit Care 2017;21(1):178.
63. Porteous J, Cioccari L, Ancona P, et al. Outcome of acetaminophen-induced acute liver failure managed without intracranial pressure monitoring or transplantation. Liver Transpl 2019;25(1):35–44.
64. Germani G, Theocharidou E, Adam R, et al. Liver transplantation for acute liver failure in Europe: outcomes over 20 years from the ELTR database. J Hepatol 2012;57(2):288–96.
65. O’Grady JG, Alexander GJ, Hayllar KM, et al. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989;97(2):439–45.
66. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology 2001;33(2):464–70.
67. Mishra A, Rustgi V. Prognostic models in acute liver failure. Clin Liver Dis 2018; 22(2):375–88.
68. Klig JE, Sharma A, Skolnik AB. Case records of the Massachusetts General Hospital. Case 26-2014. A 21-month-old boy with lethargy, respiratory distress, and abdominal distention. N Engl J Med 2014;371(8):767–73.
69. Ghannoum M, Nolin TD, Lavergne V, et al. Blood purification in toxicology: nephrology’s ugly duckling. Adv Chronic Kidney Dis 2011;18(3):160–6.
70. Garlich FM, Goldfarb DS. Have advances in extracorporeal removal techniques changed the indications for their use in poisonings? Adv Chronic Kidney Dis 2011;18(3):172–9.
71. Fertel BS, Nelson LS, Goldfarb DS. Extracorporeal removal techniques for the poisoned patient: a review for the intensivist. J Intensive Care Med 2010;25(3): 139–48.
72. Tandukar S, Palevsky PM. Continuous renal replacement therapy: who, when, why, and how. Chest 2019;155(3):626–38.